International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
| Vinod Kumar Yata*§, Anant Saxena§, Vinita Sharma§, Som Dutt§, Ajit Kumar§ Department of Biotechnology, Dr B R Ambedkar National Institute of Technology Jalandhar, Jalandhar, Punjab, India |
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
In this study, we analyzed and compared the interactions of an antibiotic drug, trimethoprim, with native and mutant human Dihydrofolate Reductase DHFR structures by computational methods. We observed that, Ile7, Glu30, Leu22, Val115, Pro61, Phe179 and Tyr182 of native DHFR play an important role in interacting with trimethoprim. Ile7, Glu30, Val112, Trp113, Ile114 and Leu133 of mutant DHFRare identified as key residues in interacting with trimethoprim. This study would help in modifying the structure of trimethoprim for improved affinity of the drug towards the native and mutant structures of human dihydrofolate reductase.
Keywords |
| Antibiotic drug, trimethoprim, dihydrofolate reductase. |
INTRODUCTION |
| Human Dihydrofolate reductase (DHFR), is an enzyme that translates dihydrofolic acid to tetrahydrofolic acid by reduction reaction using nicotinamide adenine dinucleotide phosphate [1]. DHFR has an effervescent role in regulating the amount of Tetrahydrofolate in the cell [2]. Trimethoprim (TMP) is a bacteriostatic antibiotic used mainly in the prevention and treatment of urinary tract infections [3]. TMP is a type of chemotherapeutic agent known as dihydrofolate reductase inhibitors [4]. Trimethoprim inhibits the reduction of dihydrofolic acid (DHF) to tetrahydrofolic acid (THF) by binding to dihydrofolate reductase [5]. When comparing the affinity of trimethoprim for bacterial and human dihydrofolate reductase, it is several thousand times greater than its affinity for human dihydrofolate reductase [6].Computational study of drug interactions with molecular targets helps in designing navel drugs [7,8]. In this study, we have chosen one native and one mutated structure of human DHFR for studying interactions with an antibiotic drug, trimethoprim. The mutant structure was mutated at two sites. In first mutation, glutamine (Q) at 35th position was replaced by lysine (K). In the second mutation, arginine (N) at 64th position was replaced by phenyl alanine (F). Herein, we compared the interactions of TMP with native and mutant structures (Q35K/N64F) of human DHFR by using different computational methods. |
II. EXPERIMENTAL METHODS |
| The three-dimensional (3D) structures of native (PDB ID: 1YHO) and mutant Dihydrofolate Reductase (PDB ID: 3S3V) were obtained from the crystal structures of Protein Data Bank [9, 10]. The amino acid residues binding with trimethoprim in the native and mutant structures were identified by PDBsum program [11]. The RasMol molecular graphics software is used to provide a 3D view of molecules and their interactions within PDBsum [12]. The ConSurf Server was used to compute the conservation score of binding site residues which interacted with amino acid residues in each protein [13]. The comparison of the sequence of PDB chain with the deposited proteins is calculated using this server. It also finds the ones that are homologous to the PDB sequence [14]. Residues expected to play key roles in the stabilization of proteins are selected by using Sride server [15]. |
III. RESULTS AND DISCUSSION |
Structural analysis |
| The PDB structure of both the native type (1YHO) and mutant type (3S3V) Dihydrofolate Reductase were obtained from the protein data bank and visualized using Pymol software [16]. Both the structures are complexed with trimethoprim which is showing green color while the protein as grey [figure1]. In the mutant structure of human dihydrofolate reductase (3S3V), the red and blue colors denote the mutations at position 64 and 35 respectively [figure 1B]. |
 |
Binding Residues Analysis |
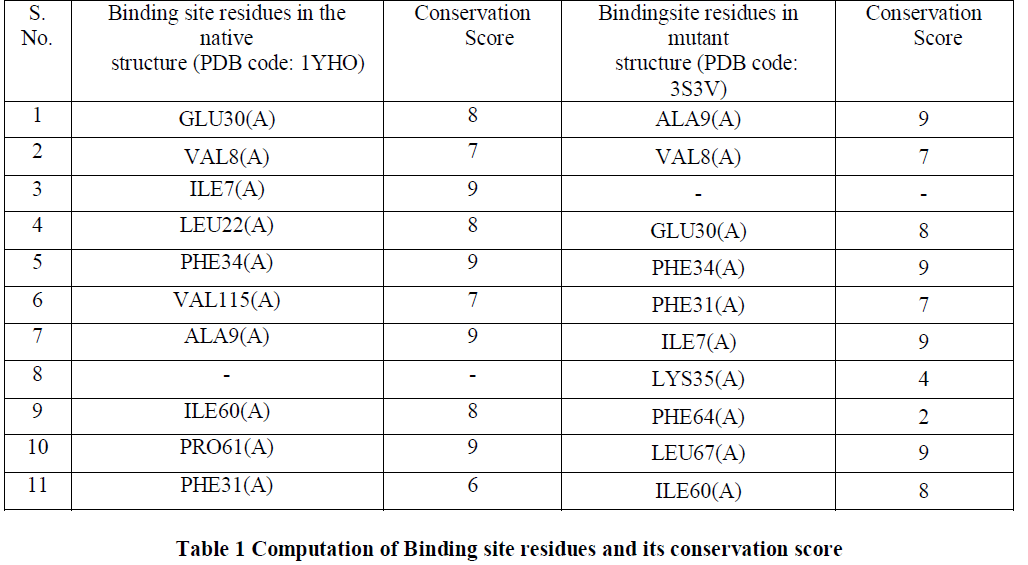
| The binding site residues for the trimethoprim in both native and mutant Dihydrofolate Reductase were identified from the PDBsum program [11] and were used for analysis and are shown in Figure2. It was observed that both the residues contain almost similar number of contacting residues. From the structures we observed that the bond length between Ile7(A) and trimethoprim was shorter in case of mutant structure (3.16 Angstrom as compared to 3.21 Angstrom of native type). Similarly the bond length between Glu30(A) and trimethoprim was also shorter in case of mutant structure (3.06Angstrom as compared to 3.78 Angstrom of native type). Shorter bond length denotes greater binding efficiency and hence we can conclude that the binding efficiency of mutant structure was better than that of native type. |
 |
Conservation Score |
| Conserved amino acid residues for both native and mutant DHFR were identified using ConSurf server [13]. The highest conservation score of 9 was achieved by 40% of the amino acid residues while 50% of the amino acid residues had a conservation score in the range of 6-8 and 10% of them had the conservation score less than 6 [Table1]. Since most of the amino acid residues involved in binding site interaction have a high conservation score, thus in our analysis we would consider the residues having the conservation score of 9 as the binding site residues. We observed that Ile7(A), Phe34(A) and Aka9(A) had the conservation score of 9 in both the native and mutant structures. Only Lys35(A) and PHE64(A) have the conservation score below 6 and they both are the residues in the mutant structure. Residues with a conservation score of 1 are considered highly variable and residues with a score of 9 are considered highly conserved residues. |
 |
Stabilizing Residues |
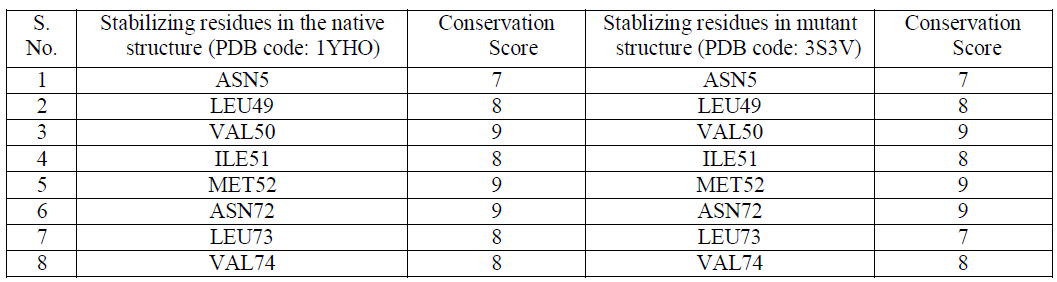
| The stability of structures was examined by calculating the number of stablizing residues by Sride server [15]. It was observed that VAL50, MET52 and ASN72 shows the highest conservation score of 9 in both the native and mutant Dihydrofolate Reductase [Table2]. Also that ILE114 and LEU134 were not observed in the native Dihydrofolate Reductase and showed a conservation score of 7 and 6 respectively. Conservation score of greater than or equal to 6 is the cut-off limit for identifying stabilizing residues. |
 |
 |
IV. CONCLUSION |
| In conclusion, we compared the interactions of trimethoprim with native and mutant structures of human DHFR by computational methods. In both the native and mutant structures trimethoprim interacts with Ile(7) and Glu(30) but the bond distances in native structure are less than in mutant structure.Leu22, Val115 and Pro61 are highly conserved in native structure but not in the mutant DHFR. In the native structure, Val115, Phe179 and Tyr182 play a key role in stabilization while in the mutant structure Val112, Trp113, Ile114 and Leu133 play an important role in the stabilization. |