Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
1Biomedical Engineering Department, Faculty of Engineering and Architecture, Kastamonu University, Kastamonu, 37100, Turkey
2Department of Forest Industrial Engineering, Faculty of Forestry, Kastamonu University, Kastamonu, Turkey
3Gebze Institute of Technology, Department of Environmental Engineering, Kocaeli, Turkey
4Department of Chemistry, Faculty of Science and Arts, Ondokuz Mayis University, Samsun, Turkey
Received date: 06/04/2016 Accepted date: 15/04/2016 Published date: 19/04/2016
Visit for more related articles at Research & Reviews: Journal of Chemistry
In this study, UV calculations of some azo-ester derivatives substituted 4-acryloyloxy group in the gas phase and the solvent phases (ethanol, dimethylformamide and chloroform) for neutral form and gas phase for their protonated forms have been studied theoretically by using density functional theory. On the UV-Vis values, bathochromic, hyperchromic or hypsochromic effects have been observed, depending on the substituents and the polarity of the media. In case of the increase of the electron donating or attractive effect to azobenzene residues, π → π* transition have been determined to undergo bathochromic shift. Result of the calculated UV spectra compared with experimental results was found almost well. Besides, results reveal that the main molecular orbitals, contributed in the electronic transitions, are highest occupied molecular orbital and lowest unoccupied molecular orbital.
Solvent effects, Computational chemistry, DFT, Electronic states, TDDFT
Azo dyes have many different structures that monoazo, diazo and triazo dyes contain one N=N double bond, two and three N=N double bonds, respectively, but comparing with other the mono azo compounds constitute the vast majority [1]. The azo groups are attached to the aromatic heterocycles, enolizable aliphatic groups and mainly benzene and naphthalene rings [2].
Azo dyes are known as the most important group in the organic colourants [3,4]. Aromatic azo compounds have been used in versatile applications in various fields because they are dominant in the synthetic colourants [5-11]. Their important applications in dying textiles and fibriles, in printing and high technology areas have been reported over the years [12]. Since azo-acrylate derivatives can be used as dyes and pigments and copolimerised with different monomers, they are commonly used in the synthetic organic fields [13,14]. Besides, The UV-Vis absorption spectra in ethanol, acetone and dimethylformamide (DMF) were studied [14].
In addition to property of high hyper-polarizability, since azobenzene derivatives are prepared for good processability by simple methods, their researches on non-linear optical materials have been broadly studied. It was resulted that introducing side chain chromophores thorough binary copolymerization may implicate chain transfer, or even retardation that is some of the unwanted occurrences [15]. This retardation means that the processability of the final products is got worse because of decreasing of molecular mass of the synthesized copolymers. New Azobenzene monomers containing methacrylate groups were synthesized in order to overcome problem [16].
Moreover, the designing of synthesis of polymers and characterization of monomers are significant for determining properties of polymers. For this reason, in the present work, we have studied theoretically molecular structure of some azoacrylate derivatives and compared with result of experimental and theoretical studies of them. In addition to molecular structure UV calculations were carried out by using the Gaussian 09 program [17].
Molecular structures of some azo-ester derivatives containing 4-acryloyloxy group were firstly optimized by using B3LYP method and 6-311G(d,p) basis set in gas phase and ethanol, DMF and chloroform phase for neutral form and gas phase for protonated form. The UV calculations for molecules 1-9 and 10-18 were secondly performed by applying Own N-layered Integrated molecular Orbital and molecular Mechanics method (ONIOM) [18]. The Kohn–Sham density functional theory (DFT) was used to calculate ground state geometries and time-dependent DFT (TDDFT) were employed to calculate excitation energies [19-21]. In this study the well-known and broadly used Becke3–Lee–Yang–Parr hybrid functional (B3LYP) with the 6-311G(d,p) basis set was used.
All compounds in this work are shown Table 1. The vertical excitation energies and oscillator strengths were obtained with TD-DFT computations at the optimized ground state equilibrium geometries using the same hybrid functional and basis set. The Self-Consistent Reaction Field (SCRF) method and the Conductor-Polarizable Continuum Model (CPCM) were used for the geometry optimizations of all the molecules in different solvent environments.
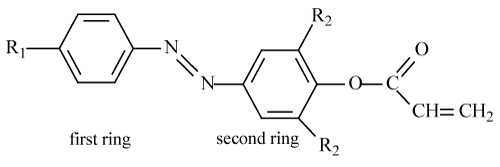 |
|||||
| Compound | R1 | R2 | Compound | R1 | R2 |
| 1 | H | H | 10 | H | CH3 |
| 2 | CH3 | H | 11 | CH3 | CH3 |
| 3 | C2H5 | H | 12 | C2H5 | CH3 |
| 4 | (CH3)3 | H | 13 | (CH3)3 | CH3 |
| 5 | NO2 | H | 14 | NO2 | CH3 |
| 6 | F | H | 15 | F | CH3 |
| 7 | Cl | H | 16 | Cl | CH3 |
| 8 | Br | H | 17 | Br | CH3 |
| 9 | OCH3 | H | 18 | OCH3 | CH3 |
Table 1: The structures of acryloiloxy derivatives.
Azo compounds have the general formula A-N=N-B where A and B indicate the aromatic structures conjugated with azo group. In this structure, in the absence of electron donating groups, azo compounds are weakly colored, and low intensity n → π* transition of the visible absorption band of the azo group are observed. If the electron-donating group is attached to A and B, intense absorption band is usually shifted to the visible region. In this situation, it is arise from that electron density shifts from donor group to chromophoric group. If D is electron donor group in the general formula D-A-N=N-B, -A-N=N-B or -N=N-B can be considered as an electron attractive group in the azo compound. Maximum bathochromic shift is observed when it is performed by connecting all of electron donating groups to A, and binding the any electron attractive group to B. This situation can be explained that -N=N-B is an attractive complex the group. Some azo compounds may contain electron donating substituents in both A and B. In some azo compounds as D-A-N=N-B type in which bathochromic effect is observed much more, π electrons can be correctly polarized in the ground state from D to B, and thus wavelength can be increased by connecting the electron attractive group to B, but hypsochromic effect is shown if it is connecting to A. The Nitro group causes more bathochromic shift than halogen groups. Bathochromic shift further increases if the electron attractive group is connected to both benzene ring, particularly in the para and orto position [22,23].
In this study, The UV-Vis spectral behaviors of the compounds were investigated theoretically solvents, absolute ethyl alcohol, chloroform and DMF. UV-Vis results are convenient with experimental results [14].
On the UV-Vis values, bathochromic, hyperchromic or hypsochromic effects were observed, depending on the substituents and the polarity of the media. In case of the increase of the electron donating or attractive effect to azobenzene residues, π → π* transition was determined to undergo bathochromic shift. Absorption wavelength of the compound, depending on the polarity of the solvent in DMF, were generally higher than those observed in the wavelength of chloroform and ethyl alcohol. We refer to as the first group in compounds 1-9, it is observed that the absorption wavelength of the azo group increases in all compounds according to the compound No. 1 that is not containing any substituent. The highest wavelength increment was observed in compounds No. 5 containing the -NO2 group which is strong electron attractive. Likewise, in the second group compound No. 10- 18, when it is compared to the compound No. 10 does not contain substituent the group, largest bathochromic shift was observed in compound No. 14. On the first ring having the variable substituent, in the para position, substituents were evaluated in two ways as the electron donor alkyl groups: (CH3, -C2H5, -C (CH3)3) halogens (-F, -Cl, -Br), methoxy (-OCH3) and electron attractive group (NO2). In the experimental results of first group compounds, it is shown that bathochromic shift effects increases by 11-12 nm in DMF for alkyl groups; 11-14 nm in chloroform; 6-9 nm in ethyl alcohol. The theoretical values are also shifted to longer wave length at range 8.3-11.1 nm for DMF; 7.8-10.6 nm for chloroform; 8.2-11.12 nm for ethyl alcohol. It is shifted to longer wavelength in the experimental results for nitro group 32 nm in DMF; 25 nm in chloroform; 15 nm in ethyl alcohol. In theoretical studies it is observed that 37.53 nm in DMF; 35.6 nm in chloroform; 37,3 nm in ethyl alcohol. In the case of the halogen substituent group is attached, in experimental results, bathochromic shifts are shown as 12-15 nm in DMF, 4-19 nm in chloroform, 3-7 nm in ethyl alcohol. In the theoretical calculation was observed an increasing of range 3.16-12.9 nm in DMF, 3.5-13.4 nm in chloroform, 3.2-12.9 nm ethyl alcohol. In the case where the methoxy group is attached, it is recorded that experimental measurements are shifted to longer wave length, 28 nm in DMF, 29 nm in chloroform 24 nm in ethyl alcohol, and in the theoretical measurements; 26.7 nm in DMF, 25 nm in chloroform, 25.8 nm in ethyl alcohol.
In the second group compounds; for bathochromic shifts of the alkyl group, experimental values is 5-12 nm in DMF, -7-0 nm in chloroform (wavelength decrease), 5-7 nm in ethyl alcohol. Theoretical values is shifted to longer wave length, 7.5-8.6 nm in DMF, 7.1-8.2 nm in chloroform, 7.4- 8.6 nm in ethyl alcohol. In the case where the nitro group is attached, experimental values is shifted to longer wave length 20 nm in DMF, 11 nm in chloroform, 19 nm in ethyl alcohol; theoretical values is shifted to longer wave length 41.2 nm in DMF, 39.2 nm in chloroform, 41 nm in ethyl alcohol. When the Halogen group substituents is bound to azobenzene, in the experimental values, it is 8-10 nm in DMF, -8-1 nm in chloroform, 2-8 nm in ethyl alcohol; In theoretical calculations, it is observed an increase at range 2.9-12.6 nm in DMF, 3.1- 13.1 nm in chloroform, 3.3-12.8 nm in ethyl alcohol. In the case where the methoxy group is attached, in the experimental measurements it is observed shifted to longer wavelength 26 nm in DMF, 7 nm in chloroform, 16 nm in ethanol; In theoretical calculations was observed shifted to longer wavelength, 24.1 nm in DMF, 22.8 nm in chloroform, 23.7 nm ethyl alcohol (Table 2).
| Gaz | Chloroform | DMF | Ethanol | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λmax (nm) | E (ev) |
F (hz) |
λmax (nm) | E (ev) |
F (hz) |
λmax (nm) | E (ev) |
F (hz) |
λmax (nm) | E (ev) |
F (hz) |
||||
| Exp | Theor | Exp | Theor | Exp | Theor | ||||||||||
| 1 | 343 | 3.62 | 0.96 | 324 | 354.01 | 3.50 | 1.08 | 331 | 353.95 | 3.50 | 1.07 | 327 | 352.49 | 3.52 | 1.06 |
| 2 | 349 | 3.55 | 1.06 | 335 | 362.06 | 3.42 | 1.17 | 343 | 362.49 | 3.42 | 1.16 | 333 | 360.95 | 3.44 | 1.15 |
| 3 | 351 | 3.54 | 1.1 | 338 | 363.01 | 3.42 | 1.20 | 343 | 362.96 | 3.42 | 1.19 | 335 | 361.99 | 3.43 | 1.18 |
| 4 | 351 | 3.53 | 1.15 | 336 | 362.82 | 3.42 | 1.25 | 342 | 362.76 | 3.42 | 1.24 | 336 | 361.36 | 3.43 | 1.22 |
| 5 | 371 | 3.34 | 1.02 | 349 | 389.71 | 3.18 | 1.13 | 363 | 390.53 | 3.17 | 1.10 | 342 | 388.82 | 3.19 | 1.09 |
| 6 | 347 | 3.57 | 0.98 | 328 | 357.53 | 3.47 | 1.08 | 343 | 357.12 | 3.47 | 1.08 | 330 | 355.68 | 3.49 | 1.06 |
| 7 | 354 | 3.50 | 1.09 | 333 | 364.35 | 3.40 | 1.20 | 346 | 363.59 | 3.41 | 1.19 | 330 | 362.16 | 3.42 | 1.18 |
| 8 | 358 | 3.46 | 1.11 | 343 | 368.25 | 3.37 | 1.22 | 343 | 367.15 | 3.38 | 1.21 | 334 | 365.73 | 3.39 | 1.20 |
| 9 | 344 | 3.61 | 0.97 | 353 | 353.76 | 3.50 | 1.07 | 359 | 352.65 | 3.52 | 1.05 | 351 | 351.28 | 3.53 | 1.04 |
| 10 | 344 | 3.60 | 0.9 | 341 | 357.39 | 3.47 | 1.04 | 332 | 357.89 | 3.46 | 1.04 | 328 | 356.43 | 3.48 | 1.02 |
| 11 | 349 | 5.23 | 0.13 | 341 | 364.26 | 3.40 | 1.15 | 344 | 365.21 | 3.39 | 1.15 | 335 | 363.65 | 3.41 | 1.13 |
| 12 | 351 | 3.53 | 1.05 | 336 | 365.34 | 3.39 | 1.18 | 337 | 366.27 | 3.39 | 1.18 | 333 | 364.76 | 3.40 | 1.17 |
| 13 | 351 | 3.53 | 1.11 | 334 | 365.23 | 3.39 | 1.23 | 339 | 366.13 | 3.39 | 1.23 | 334 | 364.67 | 3.40 | 1.21 |
| 14 | 376 | 3.30 | 0.91 | 352 | 397.32 | 3.12 | 1.04 | 352 | 399.93 | 3.10 | 1.02 | 347 | 398.27 | 3.11 | 1.00 |
| 15 | 348 | 3.57 | 0.92 | 333 | 360.62 | 3.44 | 1.05 | 340 | 360.75 | 3.44 | 1.05 | 330 | 359.32 | 3.45 | 1.03 |
| 16 | 355 | 3.49 | 1.03 | 338 | 367.55 | 3.37 | 1.17 | 342 | 367.39 | 3.37 | 1.16 | 335 | 365.95 | 3.39 | 1.15 |
| 17 | 358 | 3.46 | 1.06 | 342 | 370.83 | 3.34 | 1.20 | 340 | 370.65 | 3.35 | 1.20 | 336 | 369.20 | 3.36 | 1.18 |
| 18 | 350 | 3.54 | 0.91 | 349 | 357.22 | 3.47 | 1.04 | 358 | 357.53 | 3.47 | 1.03 | 344 | 356.15 | 3.48 | 1.02 |
Table 2: UV-Vis data of azo-ester compounds.
The UV-Vis spectral behaviors of the azo-ester derivatives containing 4-acryloyloxy groups were investigated in a variety of solvents such as absolute ethyl alcohol, chloroform and DMF by using time-dependent DFT (TDDFT) method and results was compared with the experimental values [14]. The optimized molecular structure, furthermore, the orbital shapes, highest occupied molecular orbital and lowest unoccupied molecular orbital (HOMO and LUMO), molecular electrostatic potentials (MEP) calculations, and charge distribution on atoms obtained at the B3LYP/6-311G(d,p) level for 1-18 molecules are given in Figure 1.

Figure 1: HOMO, LUMO, electron density, charge distribution and optimized of molecule 1-18.
R groups attached to molecules can increase or reduce the electron density of the ring. Electron donating groups, which are -CH3, -C2H5, C (CH3)3, F, Cl, Br, OCH3, increase the electron density of a system. Electron withdrawing groups, which is NO2, remove the electron density from the ring.
The HOMO, related directly to the ionization potential, implies the outermost orbital filled by electrons and behaves as an electron donor while the LUMO, which is directly related to the electron affinity, can be thought as the first empty innermost orbital unfilled by electron and behaves as an electron acceptor. HOMO and LUMO orbitals are named as frontier molecule orbitals (FMOs). To determine the molecular chemical stability and molecular electrical transport properties of molecules, the energy gap between HOMO and LUMO can be used [24]. The observed electronic absorption spectral results of the mentioned compounds in gas phase, ethanol, chloroform dimethylformamide solvents show the maximum wavelength values in the region 350-390 nm which can be attributed to the electronic →π* transition for studied 1-18 molecules [14]. It was observed that the transitions in the region 350-390 nm involve the promotion of one electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). From the Figure 1, it is found that the electron densities in the HOMOs and LUMO of all azoacrylate derivatives under study were largely located on the phenyl moiety and azo groups. In the studied azo- acrylate derivatives 1-18, it is observed that the energy gap of HOMO and LUMO orbitals was decreased while the solvent polarity was increased. HOMO → LUMO gap gives clear evidence for the experimental observation for the compounds 1-18.
UV visible absorption values of molecules under study was given in Table 2. The computed vertical excitation by the B3LYP method in the gas phase for the azo-ester derivatives 1-9 are 343, 349, 351, 351, 371, 347, 354, 358, 344 nm respectively and 351, 351, 376, 348, 355, 358 and 350 nm respectively for 10-18. As a result of attachment CH3, C2H5, C(CH3)3, NO2, F, Cl, Br, I groups Δλ values shifted the longer wavelengths compared without any subsistent.
It is known that dipole moment is a good measurement tool to demonstrate the asymmetry of a molecule. In this study, the highest values of the dipole moment calculation for each compound were obtained in protonated conditions. In the three solvents, probably influenced by solvent polarity, the highest value of the dipole moment is obtained in DMF (except molecule 9). When two series of compounds between 1-9 and 10-18 is analyzed, it is shown that 5 and 14 compounds containing NO2 in p-position have the highest dipole moment value. Azobenzene compounds containing halogen in the p position have second highest dipole moment value (6, 7, 8 and 15, 16, 17). If Halogens are compared among themselves, it was shown that dipole moment has decreased by ranking of p-Cl, p-F, p-Br. The lowest dipole moment is seen in p-F azobenzene due to 2sp2 hybrid orbital of C atoms that is exactly overlap with the 2p orbital of F atoms, as a result of this it may be associated with ease of electron delocalization. If there is OCH3 group in the para position, it is shown that it has a higher dipole moment according to the alkyl group and the compound not containing any substituent. When the compounds containing alkyl groups in the para position is considered in themselves, it is observed that dipole moment decreases in the following order: C2H5-, CH3-, But- (in DMF and Chloroform of first group compounds), and C2H5-, But-, CH3- (in ethanol of first group and in all solvents of second group). It is shown that 1 and 10 compounds do not containing any substituent have high dipole moment than compounds containing alkyl groups, but they have a lower dipole moment than the others (Table 3 and Figure 2).

Figure 2: The graph of dipole moment of molecule 1-18.
| Dipole Moment (Debye) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Gas | Chloroform | DMF | Ethanol | Proton | No | Gas | Chloroform | DMF | Ethanol | Proton |
| 1 | 1.59 | 1.8 | 1.86 | 1.86 | 13.31 | 10 | 1.84 | 2.23 | 2.34 | 2.34 | 12.76 |
| 2 | 1.27 | 1.44 | 1.51 | 1.5 | 14.11 | 11 | 1.31 | 1.6 | 1.7 | 1.69 | 13.5 |
| 3 | 1.42 | 1.47 | 1.54 | 1.54 | 16.08 | 12 | 1.4 | 1.69 | 1.78 | 1.77 | 15.34 |
| 4 | 1.26 | 1.42 | 1.49 | 1.48 | 19.19 | 13 | 1.32 | 1.61 | 1.71 | 1.7 | 18.33 |
| 5 | 7.32 | 8.38 | 8.67 | 8.65 | 24.54 | 14 | 7.77 | 8.97 | 9.33 | 9.3 | 23.64 |
| 6 | 2.79 | 3.15 | 3.23 | 3.22 | 16.62 | 15 | 3.2 | 3.73 | 3.88 | 3.87 | 15.96 |
| 7 | 3.45 | 3.93 | 4.05 | 4.04 | 18.92 | 16 | 3.89 | 4.53 | 4.71 | 4.7 | 18.17 |
| 8 | 3.33 | 3.79 | 3.9 | 3.89 | 22.51 | 17 | 3.77 | 4.39 | 4.57 | 4.56 | 21.6 |
| 9 | 1.13 | 1.92 | 2.21 | 2.2 | 14.2 | 18 | 1.84 | 2.32 | 2.5 | 2.48 | 13.64 |
Table 3: Dipole Moment of 1-18 Molecules.
Electron density of first phenyl ring for Molecules 1-9 is -0.071, -0.099, -0.084, -0.077, -0.021, -0.149, -0.163, -0.168, -0.079 e, respectively and those of second ring is -0.116, -0.134, -0.130, -0.187, -0.063, -0.123, -0.130, -0.142, -0.169 e, respectively. Electron density of phenyl rings for all molecules are shown in the Figure 3. As seen from the values, there are variations in the electron density of phenyl rings according to substituted groups (Table 4). When the electron densities of the molecules are analyzed, it is seen that, in the both series, the electron density of the phenyl ring of 5 and 14 of the compounds containing the NO2 group is observed to be lower than the others. Donor or electron attractive effects of the other substituents groups are consistent with the electron density values.

Figure 3: The graph of electron density of phenyl rings for molecule 1-18.
| No | 1.Phenyl | 2.Phenyl | Natural Charge | No | 1.Phenyl | 2.Phenyl | Natural Charge | ||
|---|---|---|---|---|---|---|---|---|---|
| N | O | N | O | ||||||
| 1 | -0.07105 | -0.11601 | -0.419 | -1.088 | 10 | 0.06177 | 0.00008 | -0.417 | -1.107 |
| 2 | -0.09898 | -0.13424 | -0.423 | -1.089 | 11 | 0.05871 | -0.00471 | -0.422 | -1.107 |
| 3 | -0.08418 | -0.13025 | -0.424 | -1.089 | 12 | 0.0575 | -0.00324 | -0.421 | -1.107 |
| 4 | -0.07693 | -0.18746 | -0.422 | -1.089 | 13 | 0.06514 | -0.00364 | -0.421 | -1.107 |
| 5 | -0.02095 | -0.0631 | -0.411 | -1.079 | 14 | 0.09723 | 0.02658 | -0.192 | -1.102 |
| -0.218 | |||||||||
| 6 | -0.1485 | -0.12321 | -0.422 | -1.087 | 15 | -0.00049 | 0.00038 | -0.421 | -1.107 |
| 7 | -0.16293 | -0.12977 | -0.419 | -1.085 | 16 | -0.00744 | 0.00555 | -0.418 | -1.106 |
| 8 | -0.16754 | -0.14198 | -0.419 | -1.085 | 17 | -0.00104 | 0.00637 | -0.206 | -1.106 |
| -0.211 | |||||||||
| 9 | -0.07931 | -0.16897 | -0.418 | -1.086 | 18 | 0.03395 | -0.02747 | -0.419 | -1.097 |
Table 4: Electron density of phenyl rings for Molecule 1-18.
Electronic energy values of molecules 1-18 are shown in Table 5 for gas phase, and solvent (dimethylformamide, ethyl alcohol and chloroform) phase. If two series of compounds between 1-9 and 10-18 is analyzed, it is shown that the largest value of Electronic Energy obtained from molecule 1 and 2 and the smallest value obtained from molecule 8 and 17.
| Energy (a.u) E(RB3LYP) | |||||
|---|---|---|---|---|---|
| No | Gaz | Chloroform | DMF | Ethanol | Proton |
| 1 | -838.92835140 | -838.93517449 | -838.93711491 | -838.93696980 | -839.26724970 |
| 2 | -878.25655614 | -878.26360892 | -878.26563357 | -878.26548177 | -878.59742649 |
| 3 | -917.58078817 | -917.58760378 | -917.58958051 | -917.58943227 | -917.92144881 |
| 4 | -996.22477771 | -996.23188872 | -996.23392171 | -996.23376941 | -996.56604090 |
| 5 | -1043.48366403 | -1043.49284665 | -1043.49538014 | -1043.49519175 | -1043.81397306 |
| 6 | -938.19285141 | -938.19958927 | -938.20147993 | -938.20133887 | -938.53047895 |
| 7 | -1298.55014355 | -1298.55681632 | -1298.55868228 | -1298.55854309 | -1298.88667290 |
| 8 | -3412.47061218 | -3412.47734486 | -3412.47922157 | -3412.47908159 | -3412.80733565 |
| 9 | -953.48375883 | -953.49232156 | -953.49477983 | -953.49459522 | -953.82744342 |
| 10 | -917.58453645 | -917.59191005 | -917.59395001 | -917.59379790 | -917.92655433 |
| 11 | -956.91269599 | -956.92029793 | -956.92242356 | -956.92226467 | -957.25652046 |
| 12 | -996.23671009 | -996.24429407 | -996.24641494 | -996.24625643 | -996.58057178 |
| 13 | -1074.88090108 | -1074.88855118 | -1074.89068815 | -1074.89052850 | -1075.22506018 |
| 14 | -1122.14004559 | -1122.14973825 | -1122.15234689 | -1122.15215362 | -1122.47383498 |
| 15 | -1016.84906595 | -1016.85634009 | -1016.85832191 | -1016.85817467 | -1017.18980664 |
| 16 | -1377.20639220 | -1377.21359963 | -1377.21555476 | -1377.21540964 | -1377.54606412 |
| 17 | -3491.12688455 | -3491.13414265 | -3491.13610681 | -3491.13596108 | -3491.46670573 |
| 18 | -1032.13980422 | -1032.14893277 | -1032.15149521 | -1032.15130352 | -1032.48620489 |
Table 5: Electronic Energy values of 1-18 Molecules.
The UV-Vis spectral behaviors of the azo-ester derivatives containing 4-acryloyloxy group were investigated in a variety of solvents such as absolute ethyl alcohol, chloroform and DMF with time-dependent DFT (TDDFT) method. On the UV-Vis values, bathochromic, hyperchromic or hypsochromic effects were observed, depending on the substituents and the polarity of the media. In case of the increase of the electron donating or attractive effect to azobenzene residues, π → π* transition was determined to undergo bathochromic shift.
The highest wavelength increment was observed in compounds containing the -NO2 group which is strong electron attractive. In addition, 5 and 14 compounds containing NO2 in p-position have the highest dipole moment value. Furthermore, the electron density of these compounds is observed to be lower than the others. Besides, donor or electron attractive effects of the other substituents groups are consistent with the electron density values. Moreover, it was resulted that molecule 1 and 2 have high electronic energy while molecule 8 and 17 have the low electronic energy.