Research & Reviews: Journal of Pharmaceutical Analysis
e-ISSN: 2320-0812
e-ISSN: 2320-0812
Rajan Salwan*
Department of Medical Science, All India Institute of Medical Sciences, New Delhi, India
Received: 27-Dec-2022, Manuscript No. JPA-22-84788; Editor assigned: 29-Dec-2022, PreQC No. JPA-22-84788 (PQ); Reviewed: 12-Jan-2023, QC No. JPA-22-84788; Revised: 27-Feb-2023, Manuscript No. JPA-22-84788 (R); Published: 06-Mar-2023, DOI: 10.4172/2320-0812.12.1.002
Citation: Salwan R. Carbohydrates as Building Blocks for the Synthesis of Medicinally Important Molecules. RRJ Pharm Anal. 2023;12:002.
Copyright: © 2023 Salwan R. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Research & Reviews: Journal of Pharmaceutical Analysis
A wide array of natural products are characterized by the presence of carbohydrate entities. Apart from oligo and polysaccharides, these include glycolipids and glycoproteins. Together, these glycoconjugates play a role in many different biological processes. Organic chemists are faced with the challenge to prepare suitable quantities of specific glycoconjugates, and their synthetic analogues, in order to unravel these processes. Fortunately, the monosaccharide building blocks, of which glycoconjugates are assembled, are in most cases available in large quantities and glycoconjugate synthetic studies are largely devoted to the development of efficient strategies to interconvert and oligomerise these monosaccharides. The accessibility of monosaccharides as cheap chiral starting materials that are endowed with multiple functional groups has inspired organic chemists to use them as starting material in the total synthesis of a wide range of complex natural products, compounds that, other than glycoconjugates, do not necessarily contain carbohydrate entities in their structure.
Building blocks; Monosaccharides; Carbohydrate; Glycoconjugate; Organic chemists
As such, carbohydrates are important components of the chiral pool from which organic chemists may choose their starting point. Moreover, many synthetic studies have appeared over the decades in which monosaccharides have been transformed into compounds that resemble the structure and/or function of natural carbohydrates and glyco conjugates. These carbohydrate mimics include compounds that find application as glycosidase and glycosyl transferase inhibitors in the study of the biosynthesis and processing of glycoconjugates [1].
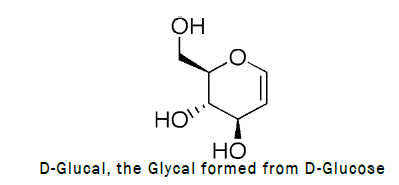
Glycal is a name for cyclic enol ether derivatives of sugars having a double bond between carbon atoms one and two of the ring. The term “glycal” should not be used for an unsaturated sugar that has a double bond in any position other than between carbon atoms one and two. Glycals can be formed as pyranose (six-membered) or furanose (five membered) rings, depending on the monosaccharide used as a starting material to synthesize the glycal. Glycals can also be classified as endo-glycals or exo-glycals. A glycal is an endo-glycal when the double bond is within the ring. If the hydroxyl group on carbon 1 has been replaced with another carbon atom, a double bond can also form outside the ring between carbon 1 and this new carbon. In this case, the product is called an exo-glycal. More recently, glycals have also been shown to be excellent starting substrates for library development, many synthetic approaches for the preparation of glycals have been devised. The original Fischer–Zach method, which uses zinc dust in acetic acid in the reductive elimination of acylated glycosyl bromides, has been one of the most popular methods for synthesizing glycals. It has been suggested that heterolytic cleavage of the carbon–halogen bond occurs under these acidic conditions, initially to give an anomeric carbocation that, after taking two electrons from the zinc atom, generates a transient carbanion that evolves through the splitting off of an acetate anion. Other reducing agents used in this transformation with glycosyl halides include sodium and potassium metal, sodium naphthalide, zinc/silver graphite, aluminum amalgam, SmI2, potassium graphite, lithium/ammonia, chromium(II), zinc/base, cobalt(II), and titanium(III).
A glucal derivative has also been prepared by introduction of a halogen atom at C-2, followed by a reductive elimination reaction in the opposite sense [2].
Synthesis of different carbohydrate based building blocks
Synthesis, protection and deprotection of D-Glucal
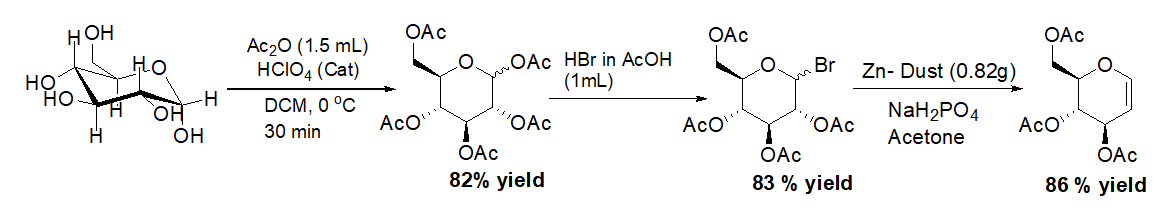
Synthesis: Synthesis of D-glucal was carried out by reacting D-glucose with different chemicals as the block diagram for the preparation of D-glucal is given below.
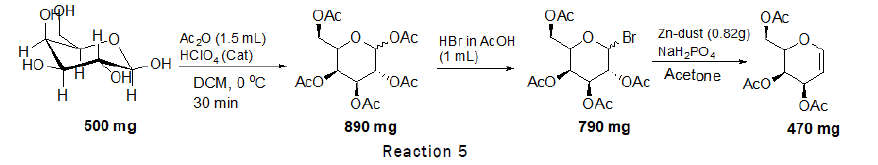
Procedure for the synthesis of D-Glucal: To a stirred solution of D-Glucose (2.77 mmol, 500 mg) in Dichloromethane (DCM), acetic anhydride (1.5 mL) and catalytic amount of perchloric acid (HClO4) was added at 0°C. The reaction wasleft for 30 minutes at 0°C. After complete consumption of starting material monitored through TLC, a solution of HBrin acetic acid (1 mL) was added drop wise at 0°C and stirred the reaction mixture at RT for 6 h. After completionquench the reaction mixture with ice cooled water and dilute with ethyl acetate. Wash the reaction mixture withsaturated solution of sodium bicarbonate twice before drying the organic layer over sodium sulphate. Collect theorganic layer dried over rotaevapourator to get crude 1-bromo-2,3,4,6-tetra-O-acetyl-D-glucose. To the crude productzinc dust (0.82 g, 12.5 mmol) and NaH2PO4 (4.6 g, 38.44 mmol) were added. The reaction mixture was stirred for 3 hat room temperature, TLC was checked (2:1 petroleum hexane–ethylacetate) indicated that the reaction wascomplete. The solution was extracted with ethylacetate. The organic phase was washed with water, saturated NaHCO3,and brine, then dried over MgSO4 and concentrated [3]. The residue was purified by silica gel column chromatography(4:1→2:1 petroleum hexane–ethyl acetate). Reaction scheme for the preparation of D-glucal is given below (Figure 1).

Figure 1: Method for the preparation of D-Glucal.
Reaction scheme
Uses of reagents:
Zn-dust: In scheme 1, it is used for dehydrohalogenation.
NaH2PO4: It is used as pH buffer
Acetone: It is used as a solvent.
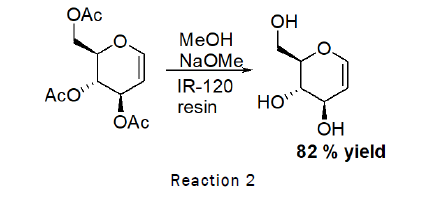
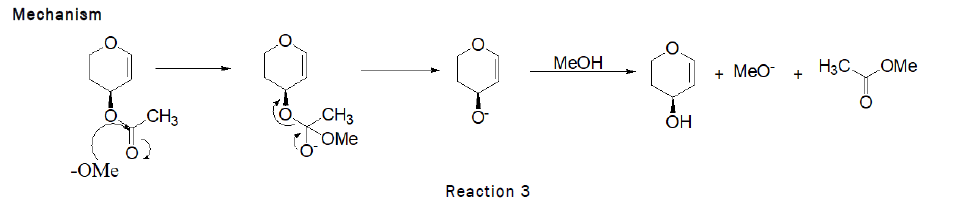
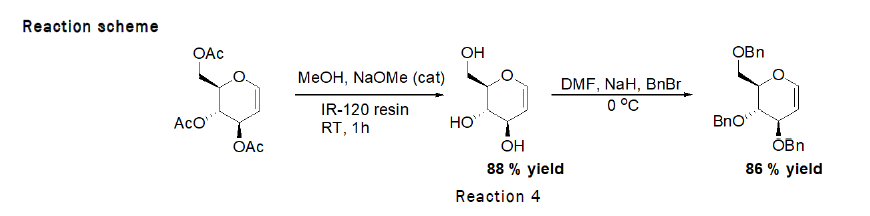
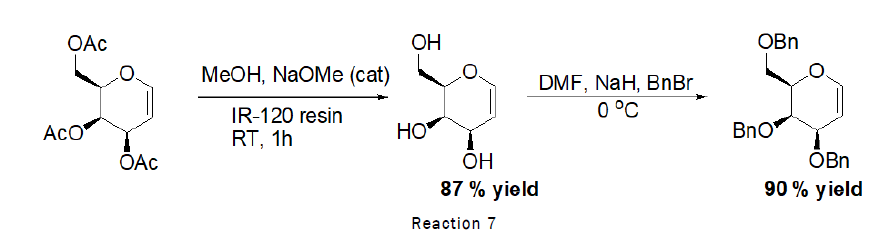
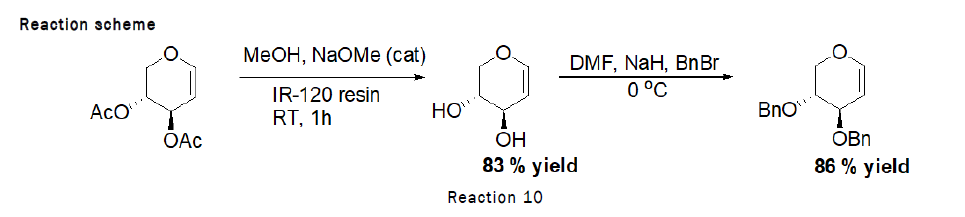
Deprotection of 3,4,6-tri-O-acetyl-D-Glucal (Zemplen Deacetylation): To the stirred solution of D-glucal (490 mg, 1.80 mmol) in methanol (1 mL), sodium methoxide (100 mg), IR-120 resin (20 mg) was added and the reaction was left for 1 h at RT. At the end TLC was checked (2:1 petroleum hexane–ethylacetate) indicated that the reaction was complete. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane–Ethyl acetate). As the reaction scheme is given below [4].
Benzylation of D-Glucal (Protection of D-Glucal): Benzylation or protection of D-glucal was carried out by reacting D-glucal with different chemicals. As the procedure of benzylation is given below.
Procedure for Benzylation of D-Glucal: To the stirred solution of 3,4,6-Tri-O-acetyl-D-glucal (500 mg, 1.83 mmol) in methanol (10 mL), catalytic sodium methoxide (118 mg, 2.196 mmol) was added at RT followed by the addition of IR 120 resin (20 mg), reaction was left for 20 min at RT. After completion, reaction mixture was separated from resin and dried over the rotaevapourator. To the stirred solution of obtained product (268 mg, 1.83 mmol) in DMF (1 mL), sodium hydride (200 mg, 8.33 mmol) was added, followed by the dropewise addition of benzylbromide (7.82 mL) at 0°C. The reaction was left for 4 h at RT (Figure 2). At the end TLC (2:1 petroleum hexane–ethylacetate) indicated that the reaction was complete. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane–ethylacetate) [5].

Figure 2: Block diagram for Benzylation of D-Glucal.
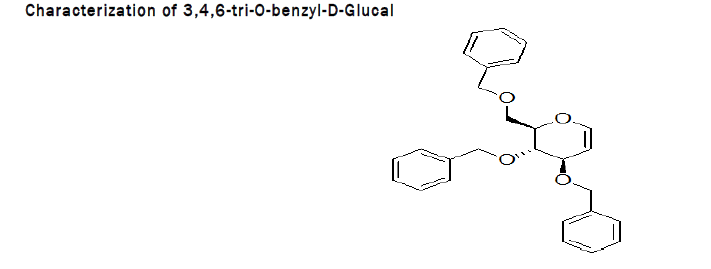
Spectral data of 3,4,6-tri-O-benzyl-D-Glucal: 1H NMR (400 MHz, CDCl3) δ 7.42–7.25 (m, 15H, Ph), 6.46 (d, J=6.1 Hz, 1H, H1), 4.95–4.82 (m, 2H, CH2), 4.68 (dd, J=11.5, 5.2 Hz, 2H, CH2), 4.61 (m, 3H, CH2, H2), 4.25 (d, J=5.6 Hz, 1H, H3), 4.11 (m, 1H, H5), 3.96–3.77 (m, 3H, H4,H6a,6b) (Figure 3).

Figure 3: NMR plot of 3,4,6-tri-O-benzyl-D-Glucal.
Characterization of 3,4,6-tri-O-acetyl-D-Glucal
Spectral data of 3,4,6-tri-O-acetyl-D-Glucal: 1H NMR (400 MHz, CDCl3) δ 6.49 (dd, J=6.2, 1.1 Hz, 1H,H5), 5.41–5.32 (m, 1H, H4), 5.25 (dd, J = 7.5, 5.7 Hz, 1H, H3), 4.87 (dd, J=6.2, 3.3 Hz, 1H, H1), 4.73 (s, 1H, H2), 4.32–4.19 (m, 2H, H8), 2.13–2.03 (m, 9H, CH3) (Figure 4).

Figure 4: 1H NMR plot of tri-O-acetyl-D-Glucal.
Synthesis, protection and deprotection of D-Galactal
Synthesis: Synthesis of D-Galactal was carried out by reacting D-galactose with different chemicals as the block diagram for the preparation of D-Galactal is given below.
Procedure for the synthesis of D-Galactal: To a stirred solution of D-Galactose (2.77 mmol, 500 mg) in Dichloromethane (DCM), acetic anhydride (1.5 mL) and catalytic amount of perchloric acid (HClO4) was added at 0°C. The reaction was left for 30 minutes at 0°C. After complete consumption of starting material monitored through TLC, a solution of HBr in acetic acid (1 mL) was added drop wise at 0°C and stirred the reaction mixture at RT for 6 h. After completion quench the reaction mixture with ice cooled water and dilute with ethyl acetate. Wash the reaction mixture with saturated solution of sodium bicarbonate twice before drying the organic layer over sodium sulphate. Collect the organic layer dried over rota evaporator to get crude 1-bromo-2,3,4,6-tetra-O-acetyl-D-galactose. To the crude product zinc dust (0.82 g, 12.5 mmol) and NaH2PO4 (4.6 g, 38.44 mmol) were added. The reaction mixture was stirred for 3 h at room temperature, at the end TLC (2:1 petroleum hexane–ethyl acetate) indicated that the reaction was complete. The solution was extracted with ethyl acetate. The organic phase was washed with water, saturated NaHCO3, and brine, then dried over MgSO4 and concentrated [6]. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane–ethylacetate). Reaction scheme for the preparation of D-galactal is given below (Figure 5).
Reaction scheme for the preparation of D-Galactal is given below

Figure 5: Method for the preparation of D-Galactal.
Reaction scheme
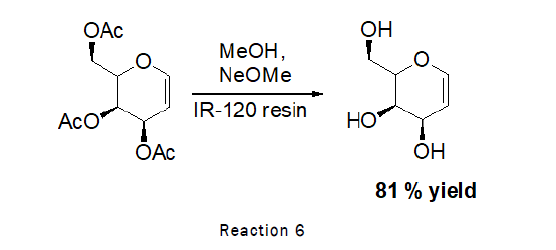
Deprotection of 3,4,6-tri-O-acetyl-D-Galactal (Zemplen deacetylation): To the stirred solution of D-galactal (470 mg, 1.72 mmol) in methanol (1 mL), sodium methoxide (100 mg), IR-120 resin (20 mg) was added and the reaction was left for 1 h at RT. At the end of which time TLC (2:1 petroleum hexane-ethylacetate) indicated that the reaction was complete. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane-ethylacetate). As the reaction scheme is given below [7].
Benzylation of D-Galactal: Benzylation or protection of D-galactal was carried out by reacting D-galactal with different chemicals. As the procedure of benzlyation is given below.
Procedure for Benzylation of D-Galactal: To the stirred solution of 3,4,6-tri-O-acetyl-D-Galactal (500 mg, 1.83 mmol) in methanol (10 mL), catalytic sodium methoxide (118 mg, 2.196 mmol) was added at RT followed by the addition of IR 120 resin (20 mg), reaction was left for 20 min at RT. After completion, reaction mixture was separated from resin and dried over the rota evaporator. To the stirred solution of obtained product (268 mg, 1.83 mmol) in DMF (1 mL), sodium hydride (200 mg, 8.33 mmol) was added, followed by the drop wise addition of benzyl bromide (7.82 mL) at 0°C. The reaction was left for 4 h at RT. At the end of which time TLC (2:1 petroleum hexane–ethyl acetate) indicated that the reaction was complete (Figure 6). The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane-ethyl acetate) [8].

Figure 6: Block diagram for Benzylation of D-Galactal.
Synthesis, protection and deprotection of D-Xylal
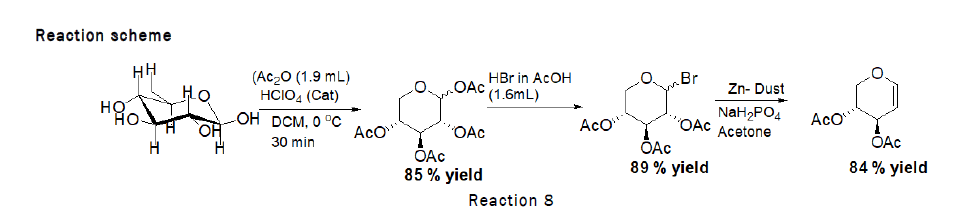
Synthesis: Synthesis of D-xylal was carried out by reacting glucose with different chemicals as the block diagram for the preparation of D-xylal is given below.
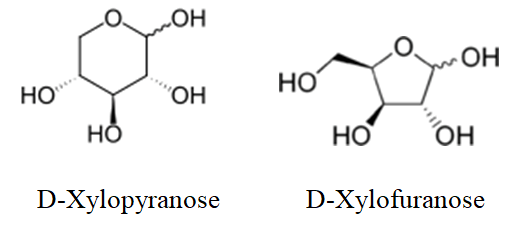
Stability: Pyranose form of glycals is more stable than the furanose form because of less angle strain as compare to furanose form. Hence mostly we get pyranose form in the product.
Procedure for the synthesis for D-Xylal: To a stirred solution of D-Xylose (3.33 mmol, 500 mg) in Dichloromethane (DCM), acetic anhydride (1.9 mL) and catalytic amount of perchloric acid (HClO4) was added at 0°C. The reaction wasleft for 30 minutes at 0°C. After complete consumption of starting material monitored through TLC, a solution of HBrin acetic acid (1.6 mL) was added drop wise at 0°C and stirred the reaction mixture at RT for 6 h. After completionquench the reaction mixture with ice cooled water and dilute with ethyl acetate. Wash the reaction mixture withsaturated solution of sodium bicarbonate twice before drying the organic layer over sodium sulphate. Collect theorganic layer dried over rota evaporator to get crude 1-bromo-2,3,4- tri-O-acetyl-D-xylose. To the crude product zincdust (0.82 g, 12.5 mmol) and NaH2PO4 (4.6 g, 38.44 mmol) were added. The reaction mixture was stirred for 3 h at room temperature, at the end TLC was checked (2:1 petroleum hexane–ethyl acetate) indicated that the reaction was complete. The solution was extracted with ethyl acetate. The organic phase was washed with water, saturated NaHCO3, and brine, then dried over MgSO4 and concentrated. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane–ethyl acetate). Reaction scheme for the preparation of D-galactal is given below (Figure 7).

Figure 7: Block diagram for the synthesis of D-Xylal.
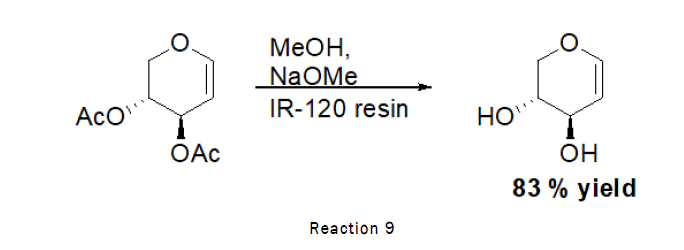
Deprotection of di-O-acetyl-D-xylal (Zemplen deacetylation): To the stirred solution of D-Xylal (440 mg, 2.2 mmol) in methanol (1.5 mL), sodium methoxide (100 mg), IR-120 resin (20 mg) was added and the reaction was left for 1 h at RT. At the end TLC was checked (2:1 petroleum hexane–ethyl acetate) indicated that the reaction was complete. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane–ethyl acetate) [9].
As the reaction scheme is given below
Benzylation of D-Xylal: Benzylation or protection of D-xylal was carried out by reacting D-xylal with different chemicals. As the procedure of benzylation is given below.
Procedure for the benzylation of D-Xylal: To the stirred solution of 3,4-di-O-acetyl-D-Xylal (500 mg, 2.5 mmol) in methanol (10 mL), catalytic sodium methoxide (90 mg, 1.66 mmol) was added at RT followed by the addition of IR 120 resin (20 mg), reaction was left for 20 min at RT. After completion, reaction mixture was separated from resin and dried over the rota evaporator. To the stirred solution of obtained product (280 mg, 1.4 mmol) in DMF (1 mL), sodium hydride (200 mg, 8.33 mmol) was added, followed by the drop wise addition of benzylbromide (7.82 mL) at 0°C. The reaction was left for 4 h at RT. At the end of which time TLC (2:1 petroleum hexane–ethyl acetate) indicated that the reaction was complete. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane–ethyl acetate) (Figure 8).

Figure 8: Block diagram for the Benzylation of D-Xylal.
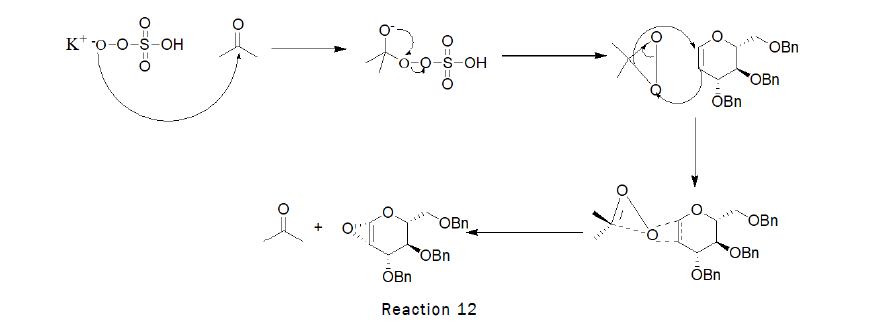
Preparation of Epoxy-O-Benzyl-D-Glucal
Procedure: To the stirred solution of 3,4,6-Tri-O-benzyl-D-Glucal (1 g, 2.40 mmol) in dichloromethane (15 mL), acetone (3 mL) was added at RT followed by the addition of saturated solution of sodium bicarbonate (30 mL),solution of oxone in water (35 mL) was added drop wise at 0°C.The reaction was left for 3 h. At the end TLC was checked (2:1 petroleum hexane–ethyl acetate) indicated that the reaction was complete [10].

Mechanism: The mechanism of epoxidation with dioxiranes most likely involves concerted oxygen transfer through a spiro transition state. As oxygen transfer occurs through the plane of the oxirane which is perpendicular to and bisects the plane of the alkene pi system. The configuration of the alkene is maintained in the product. This is syn-stereospecific reaction.
Oxone (potassium peroxymonosulphate)
Structure:
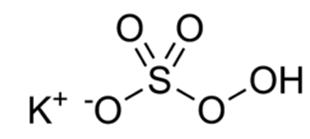
Importance: Oxone is widely used as oxidizing agent. It is the potassium salt of peroxymonosulfuric acid. This potassium salt is a component of a triple salt with the formula 2KHSO5• KHSO4• K2SO4.
Synthetic utility of sugar-enol-ethers
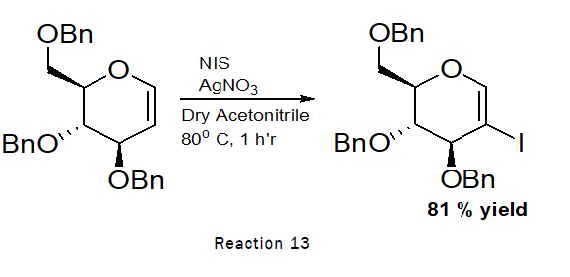
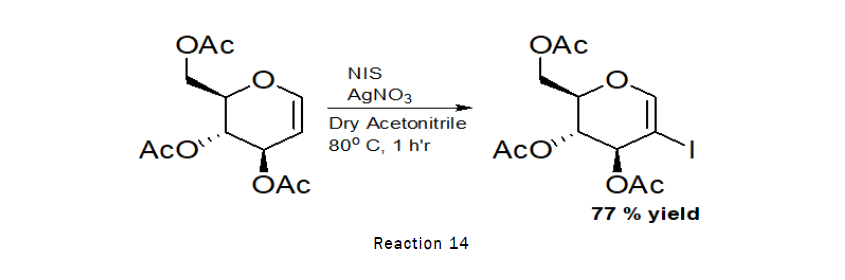
Synthesis of 2-iodo-D-Glucal
Procedure: To the stirred solution of 3,4,6-tri-O-benzyl-D-glucal (500 g, 1.20 mmol) indry acetonitrile (10 mL), N-Iodo-succinimide (500 mg, 2.23 mmol) and silver nitrate (60 mg, 0.35 mmol) was added at RT. The reaction was left for 1 h at 80°C. TLC indicated that the reaction was completed. The residue was purified by silica gel column chromatography (4:1→2:1 petroleum hexane–ethylacetate) [11].
Characterization of 2-Iodo-O-benzyl-D-Glucal
Spectral data: 1H NMR (400 MHz, CDCl3) δ 7.50–7.25 (m, 15H, Ph), 6.46 (d, J=6.1 Hz, 1H, H1), 4.97–4.82 (m, 2H, CH2), 4.72–4.65 (m, 2H, CH2), 4.60 (m, 3H, CH2, H3), 4.25 (d, J=5.6 Hz, 1H), 4.14–4.07 (m, 1H), 3.94–3.88 (m, 1H), 3.83 (dd, J=9.0, 3.9 Hz, 1H) (Figure 9).
1H NMR of 2-iodo-O-benzyl-D-Glucal

Figure 9: 1H NMR of 2-iodo-O-benzyl-D-Glucal.
Importance of sugar based Dienes
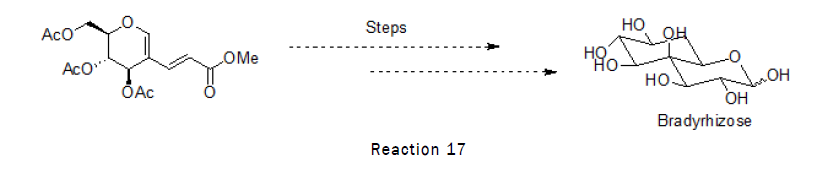
Sugar based diens are used for the synthesis of a unique Inositol fused monosaccharide Relevant to a Nod factor independent nitrogen fixation Bradyrhizose. Lipochitooligosaccharide Nod factors play a key role in the initiation of the symbiotic associations between legume plants and nitrogen fixing bacteria. Expressed by bacteria and recognized by plants, Nod factors can cause a series of biological consequences in legume root hairs to result eventually in the so called nodules that are responsible for the fixation of atmosphere N2. In 2007, Giraud, et al., discovered that Bradyrhizobium sp. BTAi1, a Gram-negative soil bacterium, was Nod factor independent in its nitrogen-fixing symbiosis with legume Aeschynomene indica. This finding disclosed a different yet unknown nitrogen fixation mechanism. Extensive studies have confirmed the necessity of LPSs in either nitrogen fixation or innate defense response. LPSs (Lipopolysaccharides) are composed of an O-antigen polysaccharide, a core oligosaccharide, and a lipid moiety. The variation of their structures is able to influence the microbe-plant interaction significantly. A structural analysis of Bradyrhizobium LPSs revealed an unprecedented O-antigen polysaccharide which consists of a structurally unique bicyclic monosaccharide named bradyrhizose [12].
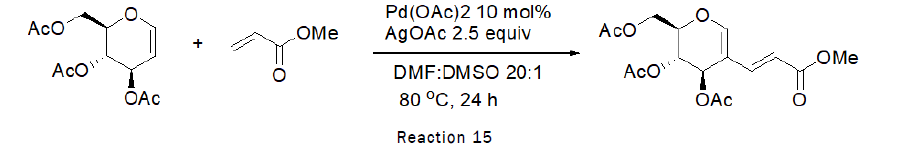
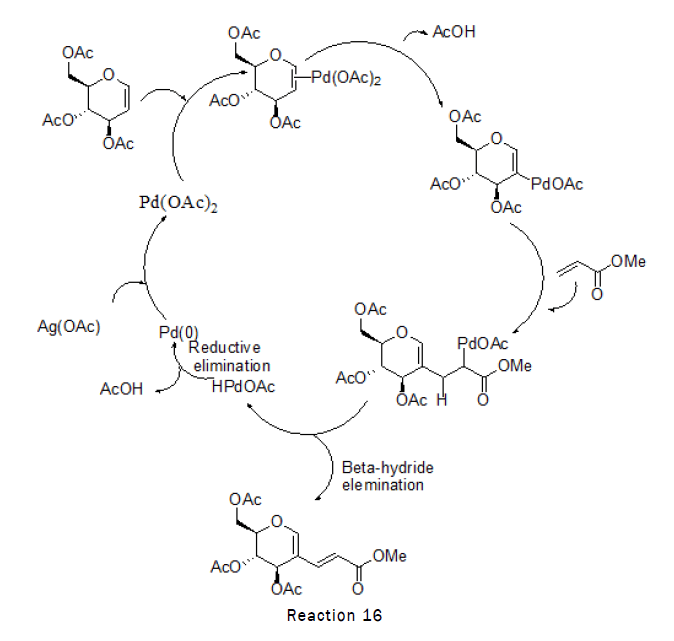
Synthesis of sugar based Dienes
Mechanism
Diene reaction scheme
Characterization of sugar based diene
Spectral data of sugar based diene: 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J=15.9 Hz, 1H), 6.92 (s, 1H), 5.57 (d, J=15.8 Hz, 1H), 5.53 (d, J=2.2 Hz, 1H), 5.10 (t, J=3.4 Hz, 1H), 4.46–4.34 (m, 2H), 4.12 (dd, J=11.6, 3.9 Hz, 1H), 3.66 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 2.02 (s, 3H) (Figure 10).
NMR

Figure 10: 1H NMR of sugar based diene.
Techniques used
Thin layer chromatography: Chromatography is a technique used to separate mixtures of compounds for either analytical identification or quantification, or for preparative purification. A mixture of compounds is dissolved in liquid and the solution (mobile phase) is transported along an immobile non-soluble adsorbent (stationary phase). Along the way, the solute molecules interact with the stationary in repetitive adsorption/desorption steps. Various compounds have degrees of interaction with the adsorbent and therefore have different mobility’s. A compound that has stronger interactions with the stationary phase will move along slower than a compound that has interaction with the adsorbent [13]. This leads to separation due to difference in mobility. Thin Layer Chromatography (abbreviated as TLC) uses a thin layer of adsorbent material (stationary phase) that is bound to a solid support (glass, plastic or aluminum sheet). The most common stationary phase is silica gel, an amorphous form of silicon oxide (SiO2) that has hydrated surfaces. Silica gel is a porous material and therefore has a very large surface area, allowing for extensive interactions with molecules. Capillary is a tube with a very small diameter. When a glass capillary is inserted into water, the water inside the capillary will rise above the level of the water outside, due to the interactions between the water molecules and the surface of the glass. Glass is mainly composed of silicon oxide which displays hydroxyl groups on its surface. The hydroxyl groups form strong hydrogen bonds with water molecules and therefore the molecules will “climb” up the glass surface to gain binding energy. The adhesion of the molecules is countered by gravity forcing the water down. As the capillary smaller, the weight of the water column inside of it decreases relative to the adhesion. When silica gel particles are packed to a thin layer, microscopic gaps exists between the particles. The gaps create a network of capillaries, and therefore once a plate is inserted into a solvent, the solvent will rise up. Therefore, TLC relies on a thin layer of adsorbent as a stationary phase and the ascending solvent as a mobile phase. A drop of solution containing a sample for separation is placed at one end of the plate. This process is termed ‘spotting’ and the spot is the origin point for the TLC. The solvent from the sample will immediately evaporate leaving behind a spot of the compound(s) adsorbed to the stationary phase. The TLC plate is then transferred to a jar containing an appropriate volume of a suitable solvent (mobile phase). The mobile phase will ascend the TLC plate (layer of adsorbent) as a result of the capillary action and it will reach the sample. The components in the sample mixture will be dissolved and carried by the ascending solvent. This is a dynamic process in which the molecules are constantly adsorbed to the surface and dissolve back in the mobile solvent. Structural differences of the compounds are manifested in the differences in reversible "binding" between the solute(s) and the binding sites on the surface of the adsorbent. Each compound in the mixture will travel a specific distance along the TLC plate leading to a series of spots. When the solvent front is close to the top of the TLC plate, the plate is then removed from the jar and the position of the solvent front is marked with a pencil. The spots on the TLC plate are analyzed either directly if the compounds are coloured or with the aid of an indicator (UV-light, or I2) if they are colorless. To identify sugars we dip The TLC plate into a solution of ethanol and 10% sulphuric acid called charring solution. Then we blow this using a hot gun. This results in if the Compounds or the components of a sample mixture differ in structure and/or in the presence, number and/or types of functional groups. Therefore, different compounds have different polarities and will vary in their capacity for interaction (intermolecular forces) with the adsorbent (stationary phase) that coats a TLC plate. As a result, different compounds will ascend a TLC plate at different rates (distances) relative to the solvent. In this way, components of a sample mixture are separated from one another, and often, one can identify the components of a mixture by comparing the distance of migration that an "unknown" travelled compare to the distance observed for "known substances" (standards). The distance that a compound travels along a TLC plate depends on few factors: The polarity of the compound (solute); the polarity of the solvent (mobile phase); and the polarity of the adsorbent (stationary phase). There is a reversible "binding" between the moving compound (solute) and the polar sites on the surface of a polar adsorbent. This "binding" is due to intermolecular forces. The same holds true for the interaction of solvent with the adsorbent. In total, there is a threefold competitive interaction between solute, solvent and adsorbent. The result of these interactions determines the relative rate (distance) at which a solute and the solvent front ascend the layer of adsorbent on the TLC plate. Thus, for a given mobile phase and adsorbent, the more polar the solute, the greater the interaction or affinity that the solute will have for the adsorbent and the less distance the solute will travel along the TLC plate. Alternatively, the less polar the solute, the weaker the interaction will be between the solute and adsorbent and more distance will be travelled by the solute in this case [14].
Rotatory evaporator
Distillation is a method of separating mixtures based on differences in volatilities of components in a boiling liquid mixture. Distillation is a unit operation, or a physical separation process, and not a chemical reaction.
A rotary evaporator (or rotavap) is a device used in chemical laboratories for the efficient and gentle removal of solvents from samples by evaporation. When referenced in the chemistry research literature, description of the use of this technique and equipment may include the phrase "rotary evaporator", though use is often rather signaled by other language (e.g., "the sample was evaporated under reduced pressure").
The main components of a rotary evaporator are:
• A motor unit that rotates the evaporation flask or vial containing the user's sample.
• A vapour duct that is the axis for sample rotation, and is a vacuum-tight conduit for the vapour being drawnoff of the sample.
• A vacuum system, to substantially reduce the pressure within the evaporator system.
• A heated fluid bath (generally water) to heat the sample.
• A condenser with either a coil passing coolant, or a "cold finger" into which coolant mixtures such as dry iceand acetone are placed.
• A condensate collecting flask at the bottom of the condenser, to catch the distilling solvent after it recondenses.
• A mechanical or motorized mechanism to quickly lift the evaporation flask from the heating bath.
The vacuum system used with rotary evaporators can be as simple as a water aspirator with a trap immersed in a cold bath (for non-toxic solvents), or as complex as a regulated mechanical vacuum pump with refrigerated trap. Glassware used in the vapour stream and condenser can be simple or complex, depending upon the goals of the evaporation, and any propensities the dissolved compounds might give to the mixture (e.g., to foam or "bump"). Commercial instruments are available that include the basic features, and various traps are manufactured to insert between the evaporation flask and the vapour duct. Modern equipment often adds features such as digital control of vacuum, digital display of temperature and rotational speed, and vapour temperature sensing [15].
Work up/down streaming: Downstream processing refers to the recovery and purification of biosynthetic products, particularly pharmaceuticals, from natural sources such as animal or plant tissue or fermentation broth, including the recycling of salvageable components and the proper treatment and disposal of waste. It is an essential step in the manufacture of pharmaceuticals such as antibiotics, hormones (e.g. insulin and human growth hormone), antibodies (e.g. infliximab) and vaccines; antibodies and enzymes used in diagnostics; industrial enzymes; and natural fragrance and flavour compounds. Downstream processing is usually considered a specialized field in biochemical engineering, itself a specialization within chemical engineering, though many of the key technologies were developed by chemists and biologists for laboratory-scale separation of biological products.
Downstream processing and analytical bio separation both refer to the separation or purification of biological products, but at different scales of operation and for different purposes. Downstream processing implies manufacture of a purified product fit for a specific use, generally in marketable quantities, while analytical bio separation refers to purification for the sole purpose of measuring a component or components of a mixture, and may deal with sample sizes as small as a single cell.
A widely recognized heuristic for categorizing downstream processing operations divides them into four groups which are applied in order to bring a product from its natural state as a component of a tissue, cell or fermentation broth through progressive improvements in purity and concentration.
Removal of insoluble is the first step and involves the capture of the product as a solute in a particulate-free liquid, for example the separation of cells, cell debris or other particulate matter from fermentation broth containing an antibiotic. Typical operations to achieve this are filtration, centrifugation, sedimentation, precipitation, flocculation, electro precipitation, and gravity settling. Additional operations such as grinding, homogenization, or leaching, required to recover products from solid sources such as plant and animal tissues, are usually included in this group.
Product isolation is the removal of those components whose properties vary markedly from that of the desired product. For most products, water is the chief impurity and isolation steps are designed to remove most of it, reducing the volume of material to be handled and concentrating the product. Solvent extraction, adsorption, ultrafiltration, and precipitation are some of the unit operations involved.
Product purification is done to separate those contaminants that resemble the product very closely in physical and chemical properties. Consequently steps in this stage are expensive to carry out and require sensitive and sophisticated equipment. This stage contributes a significant fraction of the entire downstream processing expenditure. Examples of operations include affinity, size exclusion, reversed phase chromatography, crystallization and fractional precipitation.
Product polishing describes the final processing steps which end with packaging of the product in a form that is stable, easily transportable and convenient. Crystallization, desiccation, lyophilisation and spray drying are typical unit operations. Depending on the product and its intended use, polishing may also include operations to sterilize the product and remove or deactivate trace contaminants which might compromise product safety. Such operations might include the removal of viruses or depyrogenation.
A few product recovery methods may be considered to combine two or more stages. For example, expanded bed adsorption accomplishes removal of insoluble and product isolation in a single step. Affinity chromatography often isolates and purifies in a single step.
Solvent extraction
Liquid–liquid extraction, also known as solvent extraction and partitioning, is a method to separate compounds based on their relative solubility’s in two different immiscible liquids, usually water and an organic solvent. It is an extraction of a substance from one liquid phase into another liquid phase. Liquid–liquid extraction is a basic technique in chemical laboratories, where it is performed using a separatory funnel. This type of process is commonly performed after a chemical reaction as part of the work-up.
The term partitioning is commonly used to refer to the underlying chemical and physical processes involved in liquid–liquid extraction but may be fully synonymous. The term solvent extraction can also refer to the separation of a substance from a mixture by preferentially dissolving that substance in a suitable solvent. In that case, a soluble compound is separated from an insoluble compound or a complex matrix. Solvent extraction is used in nuclear reprocessing, ore processing, and the production of fine organic compounds, the processing of perfumes, the production of vegetable oils and biodiesel, and other industries.
Column chromatography
Column chromatography is one of the most useful methods for the separation and purification of both solids and liquids. This is a solid-liquid technique in which the stationary phase is a solid and mobile phase is a liquid. The principle of column chromatography is based on differential adsorption of substance by the adsorbent.
The usual adsorbents employed in column chromatography are silica, alumina, calcium carbonate, calcium phosphate, magnesia, starch, etc., selection of solvent is based on the nature of both the solvent and the adsorbent. The rate at which the components of a mixture are separated depends on the activity of the adsorbent and polarity of the solvent. If the activity of the adsorbent is very high and polarity of the solvent is very low, then the separation is very slow but gives a good separation. On the other hand, if the activity of adsorbent is low and polarity of the solvent is high the separation is rapid but gives only a poor separation, i.e., the components separated is not 100% pure.
The adsorbent is made into slurry with a suitable liquid and placed in a cylindrical tube that is plugged at the bottom by a piece of glass wool or porous disc. The mixture to be separated is dissolved in a suitable solvent and introduced at the top of the column and is allowed to pass through the column. As the mixture moves down through the column, the components are adsorbed at different regions depending on their ability for adsorption. The component with greater adsorption power will be adsorbed at the top and the other will be adsorbed at the bottom. The different components can be desorbed and collected separately by adding more solvent at the top and this process is known as elution. That is, the process of dissolving out of the components from the adsorbent is called elution and the solvent is called is called eluent. The weakly adsorbed component will be eluted more rapidly than the other.
The different fractions are collected separately. Distillation or evaporation of the solvent from the different fractions gives the pure components. Intermolecular forces, which vary in strength according to their type, make organic molecules to bind to the stationary phase. The stronger the intermolecular force, the stronger the binding to the stationary phase, therefore the longer the compound takes to go through the column.
We successfully synthesized the glycols from different commercially available carbohydrates such as glucose, galactose and xylose and then further used the glycols for the synthesis of medicinally important molecules. The accessibility of monosaccharides as cheap chiral starting materials that are endowed with multiple functional groups has inspired organic chemists to use them as starting material in the total synthesis of a wide range of complex natural products, compounds that, other than glycoconjugates, do not necessarily contain carbohydrate entities in their structure
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]