Research & Reviews: Journal of Pharmaceutical Analysis
e-ISSN: 2320-0812
e-ISSN: 2320-0812
Javed Ahmad, Kanchan Kohli, Showkat R Mir, and Saima Amin*
Department of Pharmaceutics, Faculty of Pharmacy, Hamdard Univeristy, New Delhi 110062, India.
Received Date: 22/07/2013; Accepted Date: 03/08/2013; Published Date: 14/08/2013
Visit for more related articles at Research & Reviews: Journal of Pharmaceutical Analysis
A stability indicating liquid chromatography method was developed and validated for quantitation of aliquots of the in-vitro drug release and ex-vivo permeation study for paclitaxel in a new pharmaceutical dosage form. The reverse phase high performance liquid chromatography separation was achieved on C18 LiChrospher100 (250x4.6mm, 5 μm particle size) using a mobile phase of acetonitrile: water (60:40 v/v) at a flow rate of 1.0 mL/min. The injection volume was 20 μL and elute was analyzed with UV detector set at a wavelength 227 nm. The proposed method was validated for specificity, linearity, precision, accuracy and robustness. The calibration curve of paclitaxel was observed to be linear in the range of 0.1-100 μg/ml with r2=0.999. It was found to be simple, specific, precise, accurate, and reproducible with limit of detection and quantitation 0.026 μg/ml and 0.085 μg/ml respectively. The method was suitable for the quality control of developed paclitaxel formulation, to quantify the drug and its degradation product formed under stress conditions as well as its stability assessment under accelerated conditions used to determine shelf life.
Paclitaxel, In-vitro drug release, ex-vivo permeation study, stability
Paclitaxel (Taxane) tax-11-en-9-one,5β,20epoxy-1,2α,4,7β,10β,13α-hexahydroxy-4,10-diacetate-2-benzoate-13-(α -phenylhippurate), (Figure 1) is an important antineoplastic agent isolated from the bark of Taxus brevifolia [1]. It is effective against variety of human cancers including breast and ovarian cancer [1,2]. It is lipophilic (log P 3.5), practically insoluble in water (0.3 ± 0.02 μg/ml) and therefore has very slow dissolution rate and low oral bioavailability (<8%) [3,4]. Taxol®, the current marketed parenteral formulation of paclitaxel is associated with poor patient compliance, severe hypersensitivity reaction and rapid elimination from blood circulation [5,6]. Abraxane®, another parenteral formulation of paclitaxel is colloidal suspension of drug in human albumin. It doesn’t pose hypersensitivity but cost is comparatively high. Relative to the significantly higher cost, the efficacy of Abraxane® is only marginal, therefore lacks patient compliance [6].

Figure 1: Structure of paclitaxel
The present study projects a novel oral formulation of paclitaxel, considering its inherent problems such as poor aqueous solubility, low absorption into the gut lumen and high effluxing due to P-glycoprotein transporter. The proposed formulation is self-emulsifying in nature and readily encapsulates the drug in nanocarrier immediately when comes in contact with gastrointestinal fluid. The novel composition is formulated using biocompatible lipids and surfactants of GRAS category. It is likely to show variation in the drug release upon infinite dilution with the luminal fluid. Therefore, it is justifiable to develop a stability indicating easy, sensitive, reproducible, economical and precise method for estimation of drug content in presence of the novel excipients used in the formulation.
Though literature survey revealed many methods for analysis of paclitaxel in biological fluids like capillary electrophoresis [7], LC-MS [8] and HPLC [9,10,11]. Capillary electrophoresis and LC-MS method for analysis of paclitaxel are expensive and require sample treatment which is likely to give erroneous results due to drug loss. HPLC seems to be an overwhelming choice of analysis but for paclitaxel, its poor aqueous solubility leads poor elution through column, thus RP-HPLC seems to be an economical and sensitive method for its analysis.
Further, the method reported in literature for estimation of paclitaxel in pharmaceutical dosage forms as shown in Table 1; either has used complex gradient systems with longer analysis time or is useful for simultaneous estimation of other drugs with paclitaxel. The low peak resolution with these methods is unsuitable for routine analysis. These reported methods are likely to be incapable of estimating the very low concentration of paclitaxel in presence of our formulation excipients. Thus an easy, sensitive, reproducible, economical and precise method is required for estimation of drug content in the formulation. The proposed method has been validated as per Q1A guidelines issued by International Conference on Harmonization (ICH) [24]. Our proposed method is reported to be convenient and effective for quantitative analysis of paclitaxel in presence of different oils, surfactants and co-surfactants. Also it is found to be specific and sensitive for analysis of aliquots of In-vitro drug release, ex-vivo permeation study and stability assessment of the optimized self-emulsifying formulation in simulated gastrointestinal fluid as well as under accelerated conditions.

Table 1: Estimation of paclitaxel in different pharmaceutical formulations by HPLC
Material
Paclitaxel was provided as gift sample by Fresenius Kabi Oncology Limited, India. Caprylo caproyl macrogol-8-glyceride and Glycerol monooleate were provided by Gattefosse (Saint Priest, Cedex France). Polyoxyethylene (20) sorbitan monooleate, Polyethylene glycol, HPLC grade acetonitrile, methanol, water and other chemicals were purchased from Merck (Schuchardh, Hokenbrunn, Germany).
Equipment and chromatographic conditions
A Shimadzu model HPLC equipped with quaternary LC-10AVP pumps, a variable wavelength programmable UV detector, SPD-10AVP column oven (Shimadzu), SCL-10AVP system controller (Shimadzu), a Rheodyne injector fitted with a 20 μL loop and the Class-VP 5.032 software were used for the routine analysis of drug. Chromatography was performed with reverse phase C18 LiChrospher® 100 (250 x 4.6mm, 5 μm particle size) column at ambient conditions. The mobile phase acetonitrile: water was optimized with the flow rate of 1.0 mL/min. The injection volume was 20 μL and elute was analyzed at a wavelength of 227 nm, which represents the wavelength of maximum response for all impurities.
Preparation of stock and working solution
A stock solution of paclitaxel was freshly prepared by dissolving 10 mg of paclitaxel in 2 ml acetonitrile in volumetric flask, sonicated for about 5 min and then volume was made up to 10 ml with acetonitrile. The concentration of standard stock solution was 1000 μg/ml. Further, 1 ml of stock solution of 1000 μg/ml was taken in volumetric flask and volume was made up to 10 ml with mobile phase, acetonitrile: water mixture (50:50 v/v) to make working solution of concentration 100 μg/ml. This was used to obtain subsequent dilutions for the method validation and stability studies. All the samples were homogenized in an ultrasonic bath, filtered through a 0.22 μ membrane filter and transferred to HPLC vials before analysis.
Preparation of calibration standards
The calibration standards were prepared in the concentration range 0.1-100 μg/ml on the day of analysis. Calibration standards of concentration 0.1, 0.5, 1, 5, 10, 25, 50 and 75 μg/ml were prepared by diluting 0.01, 0.05, 0.1, 0.5, 1, 2.5, 5, 7.5 ml of working solution of concentration 100 μg/ml to 10 ml in a volumetric flask with mobile phase.
Preparation of self-emulsifying oral formulation
Paclitaxel loaded self-emulsifying oral formulation was formulated using aqueous titration method [25]. Different formulation components like oils, surfactants and co-surfactants were screened on the basis of maximum paclitaxel solubility and their proportion was further optimized through construction of pseudo-ternary phase diagram [26]. Paclitaxel containing oil solution was added in the surfactant-co surfactants system with continuous stirring and vortex mixing until the isotropic mixture was formed. The formulation was subjected to In-vitro drug release through dissolution apparatus and ex-vivo intestinal transport through everted gut sac. The aliquots collected at different time intervals were analyzed for drug content using proposed validated method.
Method validation
Method was validated as per ICH guideline [24,27,28] for different parameters such as linearity, accuracy, precision, limit of detection (LOD), limit of quantification (LOQ) and robustness. The linearity of the method was established by injecting prepared calibration standard of the drug in the concentration range of 0.1 μg ml-1 to 100 μg ml-1. Each solution was analyzed in triplicate by injecting 20 μL of solution. The intra-day precision was established by analyzing drug solutions (10, 15 and 200 μg ml-1) in triplicate on the same day. The inter-day precision of the method was determined by repeating the studies on three different days. The %RSD values were calculated to determine the intra-day and inter-day precision. The accuracy of the proposed analytical method was evaluated by adding three known concentration of the drug (50%, 100% and 150%) to the preanalyzed samples and determining the recovery of the added drug. The experiment was performed in triplicate. The robustness of the method was determined by varying the different chromatographic parameters like mobile phase composition and flow rate to determine their influence on the quantitative analysis. Limit of detection (LOD) and limit of quantification (LOQ) were determined using the slope (S) of the calibration curve and standard deviation (SD) of the blank sample using formulae-
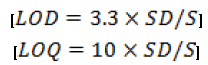
Each drug solution was analyzed in triplicate. All data was evaluated using standard statistical packages for Windows and GraphPad Prism 4.0 (GraphPad Software, Inc., USA). Statistical significance was considered at 95% probability level (p < 0.05). Pearson’s coefficient of correlation (r) was used to evaluate the significance of correlation.
Forced degradation study
The different stressed conditions applied to assess the stability of paclitaxel include:
Alkali degradation
Sample solutions of paclitaxel for alkali degradation studies were prepared in 1:1 mixture of methanol and 0.1N NaOH. The mixture was kept at 80 °C for 30 min in a water bath. The solution was allowed to attain ambient temperature and a resultant solution of 100 μg/ml concentration was analyzed by proposed method.
Acid degradation
Sample solutions of paclitaxel for acid degradation studies were prepared in 1:1 mixture of methanol and 0.1N HCl. The mixture was kept at 80 °C for 30 min in a water bath. The solution was allowed to attain ambient temperature and a resultant solution of 100 μg/ml concentration was analyzed by proposed method.
Oxidation study
Sample solutions of paclitaxel for oxidation studies were prepared in 1:1 mixture of methanol and 3% H2O2. The mixture was kept at 80 °C for 30 min in a water bath. The solution was allowed to attain ambient temperature and a resultant solution of 100 μg/ml concentration was analyzed by proposed method.
Dry heat study
Paclitaxel powder was exposed to dry heat (80 °C) in a convection oven for 8 h. Then the solution was prepared in methanol-water in 1:1 ratio to obtain 100 μg/ml concentrations and analyzed by proposed method.
Paclitaxel powder was exposed to UV light in the range 320–400 nm for 8 h to determine the effects of light irradiation on the stability of paclitaxel in the solid state. The solution of the exposed powder was prepared in 1:1 mixture of methanol-water to obtain 100 μg/ml concentrations and was analyzed by proposed method.
Stability in solution
The stability of the drug in solution during analysis was determined by repeated analysis of sample during the course of experiment on the same day and also after storage of the drug solution for 48 hour at laboratory bench conditions (25 ± 2 °C) and under refrigeration (8 ± 0.5 °C).
Stability in simulated gastrointestinal fluid
The self emulsifying formulation was diluted to a strength of 10% (v/v) and incubated at 37 °C in simulated gastric fluid (SGF) of pH 1.2 and simulated intestinal fluid(SIF) of pH 7.4, prepared according to USP specifications [29] to estimate the in vivo drug content in these fluids. Aliquots of these solutions were pipetted into glass tubes at time points 0, 15, 30 and 60 min for the gastric stability study and at 0, 1, 2 and 3 h for the intestinal stability study in triplicate. After that, samples were centrifuged then diluted 50 times with mobile phase to 10 μg/mL concentration and then filtered before HPLC analysis. The relative difference (RD) between the amount of paclitaxel added and that quantified by proposed RP-HPLC method at the end of the incubation period was calculated as follows:
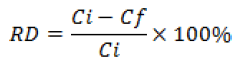
Application of the method
Paclitaxel solubility in pharmaceutical excipients
For the development of self-emulsifying oral formulation, paclitaxel solubility was determined in different formulation excipients like oils, surfactants and co-surfactants for its screening. After equilibrium solubility of paclitaxel in different excipients, supernatant was separated after centrifugation and was dissolved in methanol [26]. The dissolved drug was quantified by proposed RP-HPLC method.
In-vitro release of paclitaxel from self emulsifying formulation
Paclitaxel In-vitro release in dissolution media of pH 1.2 and pH 7.4 was done using dialysis bag technique [26,30] using USP dissolution apparatus II. Developed formulation was placed in a dialysis bag containing 2 ml of dissolution media and was securely tied and put in dissolution apparatus. 0.5 ml of dissolution sample was withdrawn at regular time interval for 8 hours and was analyzed for the drug content by proposed RP-HPLC method. The study was done in triplicate and the In-vitro dissolution profile of developed formulation was compared with aqueous suspension of pure drug as control.
Ex-vivo intestinal transport of paclitaxel from self emulsifying formulation
Paclitaxel ex-vivo intestinal transport study was done by everted gut sac method [31,32]. Everted sacs of albino wistar rat ileum were prepared using the method described by Kooshapur and Chaideh [33]. The distended gut sac was placed in 50 ml of phosphate buffer saline (PBS) of pH 7.4 containing 10 μM paclitaxel and was maintained at 37± 0.5 °C. The sac was filled with 2 ml of PBS. 200 μl of the sample solution inside the sac was taken out for quantitation of paclitaxel permeated every 15 minute till 90 minutes of the study [34]. The study was done in triplicate and ex-vivo intestinal permeation profile of developed formulation was compared with pure drug solution as control.
Accelerated stability analysis
Self emulsifying formulation was subjected to stability analysis as per protocol reported by Ivana et al, 2006, where the replicas of the formulation were kept under accelerated conditions of temperature. The replicas were stored at 30± 2 °C/75± 5%, 40± 2 °C/75 ± 5% RH and 50 ± 2 °C/75± 5% RH [35] using stability chamber maintaining the desired humidity. Replicas of the formulations were placed in the stability chamber; samples were withdrawn at a time interval of 0, 30, 60 and 90 days and analyzed for percent drug remained after appropriate dilution of the formulation with mobile phase. The amount of drug degraded and the amount remaining at each time interval were calculated. The order of degradation was determined by the graphical method. Degradation rate constant (Kdeg) was determined at each temperature using a graph plotted between log % drug remaining and time (not shown). Arrhenius plot was constructed between log Kdeg and reciprocal of temperature in Kelvin (1/T). The value of degradation rate constant at 25 °C (K25) was obtained by extrapolation of the Arrhenius plot and shelf-life was then calculated by substituting K25 in the given equation:
T0.9 = 0.1052/K25 (1)
Optimization of the chromatographic conditions
Various solvent systems were tried for the development of suitable RP-HPLC method for the quantification of paclitaxel in the bulk drug, pharmaceutical excipients and developed formulation. The selection of the mobile phase was based on sensitivity, ease of preparation, availability and suitability for drug content estimation. Paclitaxel is highly soluble in common reverse-phase HPLC mobile phase solvents, such as methanol/water and acetonitrile/water systems. But it was found that methanol in the mobile phase led to problems of high pressure and consequently acetonitrile was chosen instead. After several preliminary investigatory chromatographic runs by varying the composition of acetonitrile/water system from 40:60 to 80:20 and vice-versa, mobile phase consisting of acetonitrile: water, 60:40 v/v was optimized at a flow rate of 1 ml/min for further studies. Under the described experimental conditions, all peaks were well defined and free from tailing (Figure 2) and were detected by UV detector set at 227 nm (Figure 3). The retention time was found to be 7.5 ± 0.05 minute. The optimized process parameters are shown in Table 2.

Figure 2: Typical chromatogram of paclitaxel in the selected mobile phase acetonitrile-water (60:40)

Figure 3: UV spectrum of paclitaxel solution before and after 48 h storage at laboratory temperature 25 ± 1 °C (A) and under refrigeration 8 ± 0.5 °C (B)

Table 2: Optimized process parameters for chromatographic condition
Validation of method
Linearity
Linearity study verifies that method is able to analyze the sample solution in concentration range where analyte response is linearly proportional to concentration. This was performed by preparing calibration standard and analysis was performed in triplicate. The calibration curve between areas of peak versus concentration was found to be linear. The linear regression data for the calibration curve showed good linear relationship with respect to the peak area. The linear regression equation was found to be y = 34252x + 48913 with regression coefficient (R2) of 0.999 as shown in Figure 4. Pearson’s coefficient of correlation (r) was found to be 0.9994 with 95% confidence interval 0.9986 to 0.9997, indicative of extremely significant (P < 0.0001) linear correlation. The relative standard deviation (%RSD) of the slope prepared on three different days was 0.176%. This indicates that there is no significant difference observed in the slopes of calibration plots prepared on different days (P > 0.05).

Figure 4: Calibration plot of paclitaxel by RP-HPLC method
Accuracy
The accuracy of an analytical method is the closeness of test results obtained by that method to be true value for the sample. It is expressed as % recovery, which is determined by standard addition method. The pre analyzed samples were spiked with the extra 50, 100 and 150% of the standard, and then mixture was analyzed in triplicate by proposed method to determine the amount of drug recovered. The proposed method afforded a recovery of 99.50 - 100.73% after spiking the additional standard drug solution to the previously analyzed test solution. The results are shown in Table 3 and low value of %RSD indicates that the present method is accurate for the quantification of paclitaxel.

Table 3: Accuracy of the method
Precision
The precision of an analytical method is the degree of closeness in the determined replicate value of homogenous sample. Precision is considered at two levels of ICH suggestions i.e. repeatability and intermediate precision. Repeatability or intraday precision was carried out at three concentration level 10 μg/ml, 15 μg/ml and 20 μg/ml in triplicate on same day. While, intermediate precision or interday precision was carried out by analyzing the same concentrations of triplicate samples over three consecutive days. The results of the repeatability and intermediate precision were expressed in terms of the % RSD and are shown in Table 4. The low values of the % RSD (< 2%) indicated the repeatability of the proposed method.

Table 4: Precision of the method
Limit of detection and quantitation
The LOD and LOQ were determined by the standard deviation method. For the determination of this parameter, a blank sample was injected in triplicate. LOD and LOQ were found to be 0.026 μg/ml and 0.085 μg/ml respectively, which indicated adequate sensitivity of the method. Therefore the proposed method can be used for the detection and quantitation of paclitaxel even in very low concentrations.
Robustness
This parameter was analyzed to assess the influence of a small but deliberate variation in the chromatographic condition for the determination of paclitaxel. It was determined by introducing small changes in the flow rate (0.8 and 1.2 mL/min) and composition of the mobile phase (±2%).
There was no significant change in the retention time of paclitaxel by changing the composition of the mobile phase and flow rate. The low values of %RSD (<1%) obtained after introducing small changes in mobile phase composition and flow rate indicated robustness of the method as shown in Table 5.

Table 5: Robustness of the method
Forced degradation study
To assure the specificity of the method and to assess the stability-indicating properties of the proposed method, paclitaxel powder was stressed under various conditions. The thermolytic, photolytic, acid/alkaline hydrolytic and oxidative degradation was carried out by exposing a solution of paclitaxel to 80 °C, UV light (320-400 nm), 0.1 M HCl, 0.1 M NaOH and 3% H2O2.
The results of forced degradation studies indicated a high degree of specificity of this method for paclitaxel. Typical chromatograms obtained following the assay of stressed paclitaxel solutions are shown in Figure 5. Paclitaxel solution (100 μg/mL) stored in the refrigerator was used as a control sample for comparison purposes. The peak retention time of paclitaxel control sample was 7.5 minutes. On exposure to acid hydrolysis, oxidative degradation, elevated temperature of 80°C or exposure in UV irradiation, paclitaxel retention time was approximately 7.5 minutes in each case with a drug concentration of 72.83±0.30 μg/mL, 91.75±0.52μg/mL, 99.10±0.26 μg/mL and 98.46±0.20 μg/mL respectively as shown in Table 6. It was found that paclitaxel in solid state was stable under thermal and photolytic stressed conditions. However on exposure to alkaline hydrolysis, a cloudy gel was observed in paclitaxel solution. The resulted dispersion was filtered using 0.22 μm membrane filter and filtrate was analyzed for drug content by proposed method. It was found to be 10.33±0.48 μg/mL.

Figure 5: HPLC chromatogram of paclitaxel solution: oxidative stressed sample (A), acid hydrolysed sample (B) and alkaline hydrolysed sample (C)

Table 6: Effect of stressed conditions on pure API of paclitaxel
The structure of paclitaxel has shown in Figure 1 illustrates the ester attachment of the paclitaxel side chain to the tricyclic (baccatin) core as well as the various sites of acetylation on the diterpene core. As shown in Figure 5 (C), hydrolysis of paclitaxel in alkaline solutions results in the rapid formation of several degradants. Three early eluting degradants and a single late eluting degradant are formed upon exposure of paclitaxel to basic conditions. The elution order of the three early eluting degradants indicates they are less lipophilic than paclitaxel. Based on the structure of paclitaxel, a favorable chemical process with basic conditions would involve hydrolysis of the ester linkages. However, based on the structure of paclitaxel, a chemical process with acidic conditions would involve the addition of water to the paclitaxel core, possibly by opening of the oxetane ring. The two primary degradation products eluted prior to paclitaxel, indicate their polar characteristics versus paclitaxel as shown in Figure 5 (B). Based on the minimal degradation observed with the relatively harsh acidic conditions, paclitaxel appears to be quite stable in an acidic environment. Only a single minor degradant was observed eluting prior to paclitaxel on treatment with hydrogen peroxide solution as shown in Figure 5 (A). This degrading product has the same relative retention time as previously observed in the acid and base degraded samples. Therefore, probable degradation products can be elucidated on the basis of their chromatographic relative retention times using proposed HPLC method. Probable degradation products formed upon exposure to alkaline hydrolysis include side chain methyl ester, baccatin III, 10-deacetylpaclitaxel, and 7-epipaclitaxel. On acidic hydrolysis, probable degradation products formed are 10-deacetylpaclitaxel and the oxetane ring opened product. However, treatment with hydrogen peroxide produces only 10-deacetylpaclitaxel. The various degradation products appeared under different stressed conditions at relative retention times (RRT) of 0.19 (side chain methyl ester), 0.26 (baccatin III), 0.55 (10-deacetyl Paclitaxel), 0.81 (oxetane ring opened) and 1.21 (7-epipaclitaxel) are shown in Figure 5. The exposure of the solid drug to dry heat and photolytic stressed conditions did not result in significant decomposition. It indicated that paclitaxel in solid state is stable under thermal and photolytic stressed conditions.
The intention of this study was not to identify degradation products of drug but only to show that proposed method is specific and does not interfere, if degradation product is present in the sample of accelerated stability study.
Stability in solution
The drug solution was found to be stable for 48 hour at laboratory temperature (25 ± 1 °C) and under refrigeration (8 ± 0.5 °C) in acetonitrile-water (60:40 %v/v) as chromatogram showed no peak corresponding to the degradation products and there was no significant change in the peak area response of paclitaxel. Further, UV-spectrum also confirmed stability of drug in solution as there is no difference in shape of spectrum and λmax before and after storage of sample shown in Figure 3.
Stability in simulated gastrointestinal fluid
It gives a measure of the extent of any degradation of the drug in the presence of the gastrointestinal fluid. The extent of degradation or loss of the drug after incubation in SGF or SIF was assessed by the quantification of drug concentration by proposed method at the end of incubation time period. The relative deviation (%) of paclitaxel in SGF and SIF was no greater than 1.03 and 1.27%, respectively. Since the exposure of drug substance at 37 °C to SGF (1 h) or SIF (3 h) mimics the in-vivo drug contact with these fluids, it was concluded that paclitaxel will remain stable in the gastrointestinal milieu in developed drug carrier.
Application of the method
Paclitaxel solubility in formulation excipients
Paclitaxel solubility in different pharmaceutical excipient used for development of self-emulsifying oral formulation was conveniently quantified and results are shown in Table 7. The chromatogram of paclitaxel estimated in different pharmaceutical excipients and developed formulations are shown in Figure 6. There was no interaction observed between drug and excipients present in the self emulsifying formulation, confirming the specificity of the method.

Table 7: Solubility of paclitaxel in pharmaceutical excipients

Figure 6: HPLC chromatogram of paclitaxel in Ethanol (A), Glycerol monooleate (B), Caproyl macrogol-8-glyceride (C), Glycerol triacetate (D), Isopropyl myristate (E) and self dispersible formulation (F).
In-vitro release of paclitaxel from self emulsifying formulation
Paclitaxel In-vitro release in dissolution media at pH 1.2 and pH 7.4 was quantified using proposed method. The In-vitro dissolution profile of paclitaxel was plotted taking % cumulative amount of drug released on y-axis and time on x-axis. The profile is shown in Figure 7. The proposed method was found to be sensitive to analyze the low concentration of drug (<10 μg/ml).

Figure 7: In-vitro release profile of paclitaxel from self dispersible formulation (SDOF).
Ex-vivo intestinal permeation of paclitaxel from self emulsifying formulation
Paclitaxel ex-vivo intestinal transport was quantified using proposed RP-HPLC method. Paclitaxel permeation- time profile between cumulative amount of paclitaxel permeated and time was plotted and is shown in Figure 8. The proposed method seems to analyse nanomolar (nM) concentration of drug being absorbed through lumen.

Figure 8: Ex-vivo intestinal permeation profile of paclitaxel from self dispersible formulation (SDOF).
Accelerated stability analysis
The results of accelerated stability study illustrate slow degradation of paclitaxel at each temperature, indicating considerable stability of 50 mg paclitaxel loaded in the developed self emulsifying formulation. The order of degradation was determined by graphical method at each temperature which was found to be first order kinetics. Therefore, log % drug remaining was plotted against time and degradation rate constant (Kdeg) was calculated from the slope of the curve at each temperature as shown in Table 8. First order kinetics data was fitted to the Arrhenius equation:

Table 8: Accelerated stability analysis of paclitaxel in self dispersible formulation
Where, K is the rate constant, A the frequency factor, Ea the activation energy, R the gas constant (1.987 cal/K/mol), and T the absolute temperature. Further, the Arrhenius plot between log Kdeg values versus the reciprocal temperature (1/T × 103) in °K was found to be linear over the temperature range selected for accelerated stability study. Kinetic profile of paclitaxel in self emulsifying formulation at room temperature (25 ± 2 °C) was determined as shown in Table 9. From Arrhenius plot, the degradation rate constant at 25 °C (K25) was determined and found to be 1.2571×10-4 days-1. Further, shelf-life of paclitaxel in developed self-emulsifying formulation was calculated by substituting in equation 1 and it was found to be 2.29 years.

Table 9: Kinetic profile of paclitaxel in self dispersible formulation at 25 ± 2 °C
A convenient and rapid RP-HPLC method has been proposed for the quantitation of paclitaxel in pharmaceutical excipients used in lipid based self emulsifying formulation. In-vitro drug release and intestinal transport of paclitaxel from self-emulsifying oral formulation are accurately quantified using the proposed method. The method appears to be economical and has given better recoveries for drug in presence of interfering excipients. The developed self-emulsifying oral formulation of paclitaxel has found to be stable throughout the analysis at all temperature and humidity conditions. Thus, the proposed method can be used for routine quality control analysis of paclitaxel in lipid-surfactant based formulation development.
The authors are thankful to the University Grants Commission, Govt. of India, for funding the project work, also are thankful to Gattefosse and Nikko Chemicals for providing gift samples of surfactants and oils. The authors express their special gratitude to Fresenius Kabi oncology limited, India for providing paclitaxel as gift sample.