Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
1Pharm Technology Department, Faculty of Pharmacy, Tanta University, Tanta, Egypt
2Pharmaceutics Department, Faculty of Pharmacy Suez Canal University, Ismailia, Egypt
Received date: 08/05/2015 Accepted date: 26/06/2015 Published date: 26/06/2015
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
Diclofenac sodium (DS) is a potent non-steroidal drug with potent analgesic and anti-inflammatory activity. Its oral administration is associated with a high risk of adverse effect such as irritation, ulceration and bleeding of gastrointestinal tract. The present study focuses on the development of controlled release drug delivery system of DS. Non-aqueous emulsification solvent evaporation method was used to prepare controlled release DS particles having high DS content to obtain prolonged drug delivery in order to decrease ulcerogenicity, improve bioavailability, stability and target the drug at specific sites. Solid dispersions were prepared using EudragitRS100, EudragitRL100 and ethyl cellulose in ratios of 1:1, 1:2 and 1:3 drug to polymer. The effect of various formulations and process variables on the flowability drug loading and in-vitro drug release was studied. IR investigation revealed that there was no interaction between the drug and the polymers used. Differential thermal analysis and Powder x-ray diffractometry confirmed that the drug was reduced to molecular or microcrystalline form in the hydrophobic polymeric matrices, which could be responsible for the controlled drug release from the solid dispersions. The flow characteristic showed Hausner's ratios and Carr's index of the systems prepared indicating good and excellent flow of the systems. DS content in different solid dispersion formulations was not affected by neither the polymer type nor drug to polymer ratio and ranged between 98-100%. This study presents a new approach based on solid dispersion technique for obtaining modified release drug delivery system.
Ulcerogenicity,Diclofenac sodium controlled release drug delivery system, solid dispersion, Eudragit
Diclofenac sodium is a potent non- steroidal anti-inflammatory with analgesic and antipyretic properties. It is used for the long term therapy of chronic musculoskeletal pain, chronic inflammatory conditions, and for treatment of degenerative joint diseases such as rheumatoid arthritis, osteoarthritis, and ankylosing spondylitis [1]. DS has a short half-life (<2 h), which necessitates multiple dosing for maintaining therapeutic effect throughout the day [2-4]. For such conditions, an effective controlled release formulation would be preferred, especially if it provides an initial burst of drug to facilitate rapid onset of action and then maintain a constant plasma level for a prolonged period of time. Such a formulation will not only avoid systemic accumulation of drug and related side effects, but will also decrease dosing frequency [5].
Eudragit polymers are a series of acrylate and methacrylate polymers available in different ionic forms. Eudragit RL and Eudragit RS polymers belong to the class of poly (meth) acrylates polymers that are insoluble but permeable in digestive fluids. They possess alkaline functional groups which lend themselves to pH independent swelling (in the physiological range) and enable sustained release of active ingredient in the formulation [6,7].
Ethyl cellulose 45 CP is the ethyl ether of cellulose containing 44-51% ethoxyl (-OC2H5) groups, it is mainly used either alone or with other polymers in formulations required for making extended release preparation [8].
Non-aqueous solvent evaporation technique is a simple and highly reproducible method of enclosing or dispersing a drug in a solid dispersion. It requires only mild conditions of ambient temperature and constant stirring which enables emulsification without compromising the activity of the drug [9,10].
The main objective of this work was to investigate the possibility of obtaining a sustained release formulation of diclofenac sodium by a solid dispersion technique which was utilized by coacervation phase separation followed by solvent evaporation using Eudragit RS100, Eudragit RL100 and ethyl cellulose. Investigation of the effect of various processing and formulation factors such as drug to polymer ratio, nature of polymer on the yield production, flowability of the product as well as in-vitro release rate of the drug from the prepared solid dispersions were performed. The possibility of the presence of interaction between the drugs and polymers was determined by infrared spectral analysis, differential thermal analysis (DTA) and X-ray diffractometry.
Materials
Diclofenac sodium (Sigma- Aldrich, St. Louis, Mo, USA) was a gift sample kindly supplied by Amriya pharmaceuticals industries, Alexandria, Egypt, Eudragit RL100 and Eudragit RS 100 were purchased from RÖhm Pharma GMBH, Darmstadt (Germany), Ethyl cellulose was obtained from Sigma- Aldrich Chemi (Germany). All other reagents and chemicals were analytical grades and were used as received.
Preparation of solid dispersion
Three types of solid dispersion of Diclofenac sodium with Eudragit RL100, Eudragit RS100 and Ethyl cellulose (in ratios of 1:1, 1:2 and 1:3) drug to polymer were prepared. The content of each formula is illustrated in Table 1.

Table 1: The ratios used in solid dispersion.
The method was achieved by dissolving the calculated amount of the polymer in a mixture of ethanol: dichloride methane in a ratio (1:1) in a glass vessel at 40o C using Vortex Mixer (Maxi mix 11, Thermolyne Corporation, U.S.A.). The mixture was stirred at 400 rpm in a water bath (KOWELL N4, Germany) over 20 min. The obtained mixture was used as a solvent for the used polymers. The calculated amount of drug was gradually added to the above mixture with stirring until completely dissolved. The rotation speed of the magnetic stirrer was continued until the solvent mixture was removed by evaporation. The dry film obtained was pulverized and passed through No 450μm sieve in order to obtain a homogenous particle size [11-13]. The obtained product was kept in a desiccator over silica gel under reduced pressure until used.
Infrared spectral analysis
The IR spectrum was used to determine the interaction of the drug with the polymers used. The infrared spectra of samples were obtained using a spectrophotometer (FTIR, Jusco, Japan). Samples were mixed with potassium bromide (spectroscopic grade) and compressed into discs using hydraulic press before scanning from 4000 to 400 cm-1 [14].
Differential thermal analysis (DTA)
The physical state of drug in the solid dispersion systems was analyzed by Differential Thermal Analyzer (Mettler-Toledo star 822e system, Switzerland). The thermo grams of the samples were obtained at a scanning rate of 10°C/min conducted over a temperature range of 25-220°C, respectively [15].
X-ray diffractometry (XRD)
X-ray Diffractometry of Diclofenac sodium solid dispersion systems were performed by a diffractometer using model (Joel JDX-8030, Japan) equipped with a graphite crystal monochromator (Cu-Kα) radiations to observe the physical state of drug in the solid dispersion [15].
Flowablity testing
The flowability of the prepared powders was tested by measuring their angle of repose as well as the calculation of Carr's compressibility index.
Measuring the angle of repose
The fixed height cone method was adopted [16] where the diameter of the formed cone (d) was determined according to the following equation:
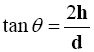
Where (h) and (d) are the height and the diameter of the cone respectively
Determination of Carr's compressibility index
A fixed weight of the powder either drug or microcapsules was poured in a 25 ml graduated cylinder, the powder was allowed to settle with no outer force and the volume occupied was measured as VI (initial bulk volume). The cylindrical graduate was then tapped on a plan surface at a one inch distance till a constant volume was obtained . The tapped volume of the powder was then recorded as (VT). The initial and tapped bulk densities were then calculated according to the following equation [17] .
Initial Bulk Density 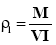
Tapped Bulk Density 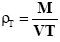
Where (M) is the mass of the powder. The percentage compressibility (Carr’s index) was then determined from the following equation:
Carr's index=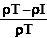
The experiments were carried in triplicate and the average angle of repose and Carr's index of each of the prepared formulae were then calculated.
Drug content determination
Percentage yield can be determined by calculating the initial weight of raw materials and the finally obtained weight of solid dispersion. Percentage yield can be calculated by using the formula:
Percentage yield= 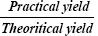 100
100
Accurately weighed drug solid dispersion were taken in a stoppered test tube and extracted with 5 × 10 ml quantities of phosphate buffer pH 7.4. The extracts were filtered and collected into 100 ml of volumetric flask and made up to the volume with phosphate buffer (pH 7.4). The solutions were subsequently diluted suitably with pre warmed phosphate buffer (pH 7.4) and spectrophotometric absorbance was taken at 276 nm [18] (UV-Visible recording spectro-photometer, SHIMADZU (UV-160A) (Japan). Percentage drug entrapment and the percentage entrapment efficiency (PEE) were calculated by the formula given below [19-21].
PEE=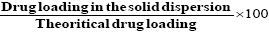
In-vitro drug release studies
The dissolution rate of diclofenac sodium solid dispersion was studied using USP dissolution test apparatus employing paddle type (Paddle type, Copley, England). Accurately weighed samples of solid dispersion were used which were calculated to contain 50 mg of the tested drug. They were placed in 900 ml of dissolution media (two types’ 0.1 N HCL (pH 1.0) and phosphate buffers (pH 7.4)). Paddle speed of 100 rpm and temperature of 37.5oC ± 0.2 was employed. Aliquots (5 ml) were withdrawn, filtered through 0.45 membrane filter and replaced with equal volume of pre warmed fresh medium to maintain constant volume and keep sink condition [22]. The drug concentration and the percentage drug released were determined with respect to time spectrophotometrically at 276 nm [22]. The in-vitro dissolution studies were performed in triplicate for each sample and the results were reported as mean ± SD.
Infrared spectral analysis
Infrared studies (Figure 1) reveal that there is no appearance of new peaks and disappearance of existing peaks, which indicated that there is no interaction between the drug and the polymers used.

Figure 1: IR spectra of pure drug Diclofenac sodium (a), Diclofenac sodium with Eudragit RS100 (b), Diclofenac sodium with Eudragit RL100 (c) and Diclofenac sodium with Ethyl cellulose (d).
The IR spectrums are shown in Figure 1. The IR spectrum of pure drug diclofenac sodium (Figure 1a) shows a characteristic peak at 3386 cm-1 due to N-H stretching frequency of secondary amine. The absorption bands at 1305 and 1282 cm-1 resulted from C-N stretching and the peaks at 1556 and 1574 cm-1 due to C=C stretching and C=O stretching of carboxylate group,respectively. The C-Cl stretching characteristic peak observed at 746 cm-1. In the IR spectra of the solid dispersions the peak due to the drug carboxyl group was shifted to 1577.49 cm−1 whereas the signal resulting from the polymer carboxyl appeared at 1734.66 cm−1 ruling out the possibility of any chemical interaction between the drug and Eudragit polymers. A weak electrostatic interaction between the carboxyl group of the drug and the ammonium group of the polymers during co-dissolution and solvent evaporation could not be ruled out.
A weak electrostatic interaction between the carboxyl group of the NSAIDs and the ammonium group of the polymer has been reported in the literature [23]. The IR spectra of diclofenac sodium pure drug (Figure 1a), diclofenac sodium with Eudragit RS100 (Figure 1b), diclofenac sodium with Eudragit RL100 (Figure 1c) and diclofenac sodium with Ethyl cellulose (Figure 1d) shows all the principal characteristic peaks related to diclofenac sodium without any change in their position, indicating no possibility of chemical interaction between the drug and formulation ingredients.
In order to confirm the physical state of the drug in the solid dispersion system, DTA of the drug alone, and drug in solid dispersion systems were carried out (Figure 2a-2d).

Figure 2: DTA thermogram of pure drug diclofenac sodium (a), diclofenac sodium with Eudragit RS100 (b), diclofenac sodium with Eudragit RL100 (c) and diclofenac sodium with Ethyl cellulose (d).
The DTA thermogram of the drug depicts a sharp endothermic peak at 280° corresponding to the melting transition temperature of diclofenac sodium [24]. The drug endothermic peak was suppressed in the thermogram of the solid dispersion suggesting that the drug was able to dissolve partially in the polymer to form a solid-solid solution. The appearance of low intensity endothermic peak also indicated the some of the drug still managed to crystallize out from the saturated Eudragit matrices during coevaporation.
X-ray diffractometry (XRD)
In order to confirm the physical state of the drug in the solid dispersion systems, powder X-ray diffraction studies of the drug alone and drug in solid dispersion systems were carried out (Figure 3a-3d). The crystalline nature of the drug was clearly demonstrated by the characteristic PXRD pattern with peaks appearing at 6.38, 8.26, 14.90, 19.23, 20.74, 25.96, 26.82 and 27.70 2θ values. Signals at 13.04, 20.96, 25.96 and 31.70 2θ values attributable to the crystalline drug appeared in the x-ray diffractogram of the solid dispersion confirmed the existence of the drug probably in the microcrystalline form in the polymer matrix. Micro-crystallization of NSAIDs from the Eudragit matrices when the drug concentration exceeded its solubility in the polymer matrix has been cited in literature [25].

Figure 3: X-ray diffractogram of pure drug diclofenac sodium (a), diclofenac sodium with Eudragit RS100 (b), diclofenac sodium with Eudragit RL100 (c) and diclofenac sodium with Ethyl cellulose (d).
The DSC and PXRD studies collectively indicated that a portion of the drug incorporated existed as solid-solid solution in the polymer matrix whereas the excess drug crystallized in the microcrystalline form from the polymeric matrices. The presence of the drug as a solid-solid solution in the hydrophobic polymeric matrix could be responsible for the controlled drug release from the solid dispersions. Solid solutions in hydrophobic polymers are responsible to prolong the drug release from multiparticulate systems [26].
Flowablity testing
The effect of presence of diclofenac sodium in solid dispersion systems on the flow properties of the drug is illustrated in Table 2.

Table 2: Flowablity test of Diclofenac sodium and its solid dispersion form.
Table 2 shows the results of the Flowablity testing of Diclofenac sodium as well as the drug with Eudragit RS100, Eudragit RL100 and Ethyl cellulose it is clear that; Carr's index for the drug alone was found to be 54.22 ± 0.65% indicating extremely poor flow. The solid dispersion systems based on Eudragit RS 100 showed an index between13.22 ± 0.13and 16.16 ± 0. 37%, while those based on Eudragit RL100 showed an index between 13.17 ± 0.56 and 16.08 ± 0.34%. The solid dispersion systems based on Ethyl cellulose showed an index between 15.43 ± 1.02 and 17.45 ± 0.49% indicating excellent and good flow [27]. In all formulations, the ratio of 1:3 drug to polymer showed the best flowability results.
It is evident from Table 2 that the angle of repose decreases by inclusion of the drug in solid dispersion systems. Also by increasing the ratio of the polymers, the angle of repose decreases i.e. Flowablity increases.
Drug content determination
Table 3 shows the results of Diclofenac sodium content in different solid dispersion systems; it is clear that the percentage yield of different solid dispersion systems varied from 98.94 ± 1.45% to 101.45 ± 1.54%. From the results in the Table it is evident that drug to polymer ratio did not play any rule in the entrapment efficiency of the drug. This is in contrast to the results obtained by Trivedi et al. [28] who reported that by increasing the polymer ratio in certain formulations from 1:1 to 1:5 was followed by increasing the drug entrapment efficiency.
Swetha et al. [29] prepared micro sponges containing etodolac with different types of polymers including Eudragit and ethyl cellulose. The authors proved that the ratio of the polymer in the delivery system has no effect on the percentage entrapment efficiency Table 3.

Table 3: Diclofenac sodium content in different solid dispersion systems in phosphate buffer (pH7.4).
In-vitro drug release studies
The release profile of Diclofenac sodium solid dispersion systems from different drug to polymer ratios at different values are shown in Figures 4-6.

Figure 4: Release of Diclofenac sodium from its solid dispersions with Eudragit RS 100, (a) in 0.1 N HCl (pH 1.0), (b) in phosphate buffer (pH 7.4).

Figure 5: Release of Diclofenac sodium from its solid dispersions with Eudragit RL 100, (a) in 0.1 N HCl (pH 1.0), (b) in phosphate buffer (pH 7.4).

Figure 6: Release of Diclofenac sodium from its solid dispersions with Ethyl cellulose, (a) in 0.1 N HCl (pH 1.0), (b) in phosphate buffer (pH 7.4)
A poor drug release ranging from 2.44 ± 0.38 to 5.18 ± 0.58 was observed from the solid dispersion systems in pH 1.0 by the end of 2 h of dissolution. This can be attributed to the pH dependent solubility of the drug, which is reported to increase at pH values higher than the pKa (4.0) of the drug [30].
The drug release from the solid dispersions in pH 7.4 depended on the total polymer levels. Solid dispersions produced with Eudragit RS alone showed slow drug release ranging from 59.72 ± 0.45% to73.21 ± 0.23% by the end of 12 h of dissolution at low and high polymer levels respectively. The amount of the drug released was inversely proportional to the polymer ratio in the solid dispersion systems.
At pH 1.0, the percentage released after 120 minutes of diclofenac in Eudragit RS100 solid dispersion system at drug to polymer ratio of (1:1) was decreased significantly (p value <0.5) than free drug at the same pH this is because Eudragit RS100 does not dissolve in acidic pH.
At pH of 7.4, the percentage released after 12 hours of diclofenac in Eudragit RS100 solid dispersion system at drug: polymer ratio of (1:1) showed significant decrease in percentage released of diclofenac than free drug at the same pH (p value <0.5).
By increasing the ratio of the polymer to decrease the percentage released of diclofenac at low pH values of 1.0 to reach (1:2) drug to polymer ratio, the obtained results showed that, At pH 1.0, the percentage released after 120 minutes of diclofenac in Eudragit RS100 solid dispersion system at drug to polymer ratio of (1:2) was decreased significantly (p value <0.5) than free drug at the same pH, and also significant (p value <0.5) than that of diclofenac in Eudragit RS100 solid dispersion system in the ratio of (1:1).
At pH of 7.4, the percentage released after 12 hours of diclofenac in Eudragit RS100 solid dispersion system at drug to polymer ratio of (1:2) was decreased significantly (p value <0.5) than free drug at the same pH, but not significant (p value >0.5) than that of diclofenac in Eudragit RS100 solid dispersion system in the ratio of (1:1).
By increasing the ratio of the polymer to decrease the percentage released of diclofenac at low pH values of 1.0 to reach (1:3) drug to polymer ratio, the obtained results showed that, At pH 1.0, the percentage released after 120 minutes of diclofenac in Eudragit RS100 solid dispersion system at drug to polymer ratio of (1:3) was decreased significantly (p value <0.5) than free drug at the same pH, and also significant (p value <0.5) than that of diclofenac in Eudragit RS100 solid dispersion system in the ratios of (1:1) and (1:2) respectively.
At pH of 7.4, the percentage released after 12 hours of diclofenac in Eudragit RS100 solid dispersion system at drug to polymer ratio of (1:3) was decreased significantly (p value <0.5) than free drug, and also significant (p value <0.5) than that of diclofenac in Eudragit RS100 solid dispersion systems in the ratios of (1:1) and (1:2) respectively at the same pH.The slow drug release from solid dispersions with Eudragit RS can be attributed to the low permeability of the polymer, which posed a significant hindrance to fluid penetration and passive drug diffusion (Figure 4).
Incorporation of the Eudragit is reported to enhance the drug release from the solid dispersion [31]. The drug release from the solid dispersions in pH 7.4 depended on the total polymer levels. Solid dispersions produced with Eudragit alone showed slow drug release ranging from 60.33 ± 0.98% to74.22 ± 0.11% by the end of 12 h of dissolution at low and high polymer levels respectively. The amount of the drug released was inversely proportional to the polymer ratio in the solid dispersion systems.
At pH 1.0, the percentage released after 120 minutes of Diclofenac in Eudragit RL100 solid dispersion system at drug to polymer ratio of (1:1) was decreased significantly (p value <0.5) than free drug at the same pH this is because Eudragit RL100 does not dissolve in acidic pH.
At pH of 7.4, the percentage released after 12 hours of diclofenac in Eudragit RL100 solid dispersion system at drug to polymer ratio of (1:1) showed significant decrease in percentage released of diclofenac than free drug at the same pH (p value <0.5).
By increasing the ratio of the polymer to decrease the percentage released of diclofenac at low pH values of 1.0 to reach (1:2) drug: polymer ratio, the obtained results showed that, at pH 1.0, the percentage released after 120 minutes of diclofenac in Eudragit RL100 solid dispersion system at drug to polymer ratio of (1:2) was decreased significantly (p value <0.5) than free drug at the same pH, but not significant (p value >0.5) than that of diclofenac in Eudragit RL100 solid dispersion system (1:1).
At pH of 7.4, the percentage released after 12 hours of diclofenac in Eudragit RL100 solid dispersion system at drug to polymer ratio of (1:2) was decreased significantly (p value <0.5) than free drug at the same pH, but not significant (p value >0.5) than that of diclofenac in Eudragit RL100 solid dispersion system (1:1).
By increasing the ratio of the polymer to decrease the percentage released of diclofenac at low pH values of 1.0 to reach (1:3) drug to polymer ratio, the obtained results showed that, At pH 1.0, the percentage released after 120 minutes of diclofenac in Eudragit RL100 solid dispersion system at drug to polymer ratio of (1:3) was decreased significantly (p value <0.5) than free drug at the same pH, and also significant (p value <0.5) than that of diclofenac in Eudragit RL100 solid dispersion system in the ratios of (1:1) and (1:2) respectively.
At pH of 7.4, the percentage released after 12 hours of diclofenac in Eudragit RL100 solid dispersion system at drug to polymer ratio of (1:3) was decreased significantly (p value <0.5) than free drug, and also significant (p value <0.5) than that of diclofenac in Eudragit RL100 solid dispersion systems in the ratios of (1:1) and (1:2) respectively at the same pH.
This increase in percentage released compared with Eudragit RS was due to increased permeability and hydrophilicity of Eudragit RL because of the higher content of quaternary ammonium group in Eudragit RL (Figure 5).
The drug release from the solid dispersions in pH 7.4 depended on the total polymer levels. Solid dispersions produced with Ethyl cellulose alone showed slow drug release ranging from 60.33 ± 0.98% to74.22 ± 0.11% by the end of 12 h of dissolution at low and high polymer levels respectively. The amount of the drug released was inversely proportional to the polymer ratio in the solid dispersion systems. This may be due to increase in system viscosity with increase in Ethyl cellulose concentration [32,33].
At pH 1.0, the percentage released after 120 minutes of diclofenac in Ethyl cellulose solid dispersion system at drug to polymer ratio of (1:1) was decreased significantly (p value <0.5) than free drug at the same pH this is because Ethyl cellulose does not dissolve in acidic pH.
At pH of 7.4, the percentage released after 12 hours of diclofenac in Ethyl cellulose solid dispersion system at drug to polymer ratio of (1:1) showed non-significant decrease in percentage released of diclofenac than free drug at the same pH (p value >0.5).
By increasing the ratio of the polymer to decrease the percentage released of diclofenac at low pH values of 1.0 to reach (1:2) drug to polymer ratio, the obtained results showed that, At pH 1.0, the percentage released after 120 minutes of diclofenac in Ethyl cellulose solid dispersion system at drug to polymer ratio of (1:2) was decreased significantly (p value <0.5) than free drug at the same pH, and also significant (p value <0.5) than that of diclofenac in Ethyl cellulose solid dispersion system in the ratio of (1:1).
At pH of 7.4, the percentage released after 12 hours of diclofenac in Ethyl cellulose solid dispersion system at drug to polymer ratio of (1:2) was decreased significantly (p value <0.5) than free drug at the same pH, but not significant (p value >0.5) than that of diclofenac in Ethyl cellulose solid dispersion system in the ratio of (1:1).
By increasing the ratio of the polymer to decrease the percentage released of diclofenac at low pH values of 1.0 to reach (1:3) drug to polymer ratio, the obtained results showed that, at pH 1.0, the percentage released after 120 minutes of diclofenac in Ethyl cellulose solid dispersion system at drug: polymer ratio of (1:3) was decreased significantly (p value <0.5) than free drug at the same pH, and also significant (p value <0.5) than that of diclofenac in Ethyl cellulose solid dispersion system (1:1) but not significant (p value >0.5) than that of diclofenac in Ethyl cellulose solid dispersion system in the ratio of (1:2) at the same pH.
At pH of 7.4, the percentage released after 12 hours of diclofenac in Ethyl cellulose solid dispersion system at drug to polymer ratio of (1:3) was decreased significantly (p value <0.5) than free drug, and also significant (p value <0.5) than that of diclofenac in Ethyl cellulose solid dispersion systems in the ratios of (1:1) and (1:2) respectively at the same pH (Figure 6).
Screening the previous results of drug release from Eudragit RS100, Eudragit RL100 and Ethyl cellulose solid dispersion systems in all ratios of (1:1, 1:2 and 1:3) at different pH values of 1.0 and 7.4, It was found that Eudragit RS100, Eudragit RL100 and Ethyl cellulose have better enteric properties especially at a ratio of (1:3). This indicates that Eudragit RS100, Eudragit RL100 and Ethyl cellulose have sufficient thickness and uniformity to prevent drug release in the gastric fluid, so that, these polymers are capable of protecting the drug in a better manner, also, increase in the drug to polymer ratios accounted for significant difference in decreasing the amount of drug released especially at low pH values of stomach.
For enteric polymers as Eudragit RS100, Eudragit RL100 and Ethyl cellulose at drug to polymer ratio of (1:1), the polymer is unable to coat the drug completely. By increasing the drug to polymer ratio to (1:3), Eudragit RS100, Eudragit RL100 and Ethyl cellulose showed significant decrease in drug release measured by percentage released after 120 minutes of diclofenac from Eudragit RS100, Eudragit RL100 and Ethyl cellulose solid dispersion systems than (p value <0.5) than free drug at the stomach pH.
There was no significant decrease in drug release from Eudragit RS100, Eudragit RL100 and Ethyl cellulose at alkaline pH of the intestine (p value >0.5).
Controlled release formulations of diclofenac sodium were successfully prepared using Eudragit RS 100, Eudragit RL 100 as well as Ethyl cellulose by non-aqueous emulsification solvent evaporation method. The in vitro release data showed controlled release of the formulation up to 12 hours. FT-IR studies did not reveal any significant drug interactions with polymers used. Differential thermal analysis and Powder x-ray diffractometry confirmed that the drug was reduced to molecular or microcrystalline form in the hydrophobic polymeric matrices, which could be responsible for the controlled drug release from the solid dispersions. Diclofenac sodium content in different formulations wasn't affected by neither the polymer type nor drug to polymer ratio. Therefore, it possible to overcome the problem of gastric damage during the use of diclofenac sodium, by avoiding the exposure of the drug to the ulcer-prone area of the GI tract by providing controlled release drug delivery systems.