Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
Hebah M Abdel-Wahab* and Joseph W Bozzelli
Department of Chemistry and Environmental Science, New Jersey Institute of Technology, New Jersey, United States
Received: 16-Jun-2022, Manuscript No. JCHEM-22-66743; Editor assigned: 20-Jun-2022, Pre QC No. JCHEM-22-66743 (PQ); Reviewed: 05-Jul-2022, QC No. JCHEM-22-66743; Revised: 17-Aug-2022, Manuscript No. JCHEM-22-66743 (R); Published: 24-Aug-2022, DOI: 10.4172/2319-9849.11.7.007
Visit for more related articles at Research & Reviews: Journal of Chemistry
Structures and thermochemical properties of Tri-, Tetra-, and Penta-Fluorinated Ethanol’s and its Radicals were determined by the Gaussian M-062 x/6-31+g (d,p) calculation : Enthalpies of formation for 18 fluorinated ethanol and some radicals were calculated with a popular ab initio and density functional theory methods: the Gaussian M-062 x/6-31+g (d,p) via several series of isodesmic reactions. Bond dissociation energies for these fluorinated ethanol’s and its radicals were also calculated.
Thermochemical; Dissociation; Nucleophiles; Epoxidation; Organic synthesis; Electrophilic reactions; Hydrocarbons
Fluorinated alcohols are known to be used in the organic synthesis industry. They have strong hydrogen bonding donor character, and they are strong nucleophiles which allow organic reactions to occur without the using of a catalyst. Fluorinated alcohols have been used as solvents in epoxidation reactions, annulation reactions, nucleophilic substitution reactions, electrophilic reactions, and functionalization of multiple bonds [1].
Fluorinated alcohols are excellent solvents of proteins, peptides, and other organic compounds due to its physicochemical properties. Fluoro alcohols, like other alcohols, can alter lipid bilayer properties and stability, and protein function [2].
Most Halogenated hydrocarbons don’t exist naturally in the environment and are synthetically produced. The major source of halogenated hydrocarbons to the atmosphere is agriculture. Crop spraying introduces halogenated hydrocarbons to the environment entering through adsorption and deposition onto airborne particles or directly entering the aquatic system. Some halogenated hydrocarbons, PCBs, furans, and dioxins are by products, industrial waste that would unintentionally enter the atmosphere [3].
The onset a temperature for energetic materials in the calorimetric measurements has been roughly predicted using molecular orbital calculations of bond dissociation energies.
Stability and reactivity of chemical compounds can be explained using bond dissociation energies values. Standard enthalpies of formation estimated using semi empirical MO calculations, MOPAC-PM7 package has been used previously to derive bond dissociation energy values for chemical compounds [4].
Bond Dissociation Energies (BDEs) values for some inorganic compounds, lanthanide selenides and sulfides were measured in 2021 using resonant two photon ionization spectroscopy. The predissociation thresholds were found to be the BDE values for these molecules. The 0K gaseous heat of formation, ΔfH°K for each molecule was also reported using this method [5].
The amount of energy used to break a mole of covalently bonded gas molecule to pair of radicals is the bond dissociation energy. The units used for the bond dissociation energy is commonly kJ/mol. Covalent bonds can be broken heterolytically or homolytically. The heterolytic breaking of a covalent bond would result in the pair of electrons going to only one atom, either A or B.
C−D→C+ + D: − or C−D→C:− + D+. The homolytic breaking of a covalent bond on the other hand would result in one electron staying with each atom,.A-B→A•+B•. Bond dissociation energy can be calculated for molecules as the difference in enthalpy of formation of products and reactants. Bond dissociation energy is a state function, as it doesn’t depend on the mechanism or pathway on how bonds form or break. Energy of chemical reactions can be assessed using values for the bond dissociation energy. There are some systematic trends for the bond dissociation values; bonds dissociation energy varies with hybridization. For example, sp3 hybridized carbons in hydrocarbons have smaller bond dissociation values compared to sp2 hybridized carbons. The longer and weaker sp3 hybridized bonds are easier to break compared the shorter and the stronger sp2 and sp hybridized bond (double and triple bonds). Among sp3 hybridized bonds, bond dissociation values depend on its position, where it’s on a primary, secondary or tertiary carbon. Methane has the strongest C-H bond with highest bond dissociation values, following C-H bonds on primary carbons, following C-H bonds on secondary carbons, following C-H bonds on tertiary carbons [6].
Energetics of chemical processes can be assessed using bond dissociation energy values. Hess’s Law has been used in the past and is currently being used to estimate reaction enthalpies by combining bond. Dissociation energies of bonds formed and bond dissociation energies of bonds broken. The energy change when forming a mole of compound from its component elements is called the enthalpy of formation, ΔHf. If heat is released when the elements combine to form the compound, enthalpy of formation would have a negative sign. If heat is absorbed when the elements combine to form the compound, enthalpy of formation would have a positive sign. A value of Enthalpy of formation is dependent upon temperature, pressure and physical states of reactants and products in the chemical reaction. Standard enthalpy of formation (ΔH°f,) is the enthalpy of formation at standard conditions; 1 atm pressure, 25°C, and 1 M aqueous solution concentration. Any element in its most stable form has a standard enthalpy of formation has a value of zero. Tabulated enthalpy of formation values can be used to calculate standard enthalpy of any reaction who’s standard enthalpy of formation values are well known [7].
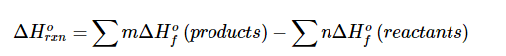
The heat involved in chemical or physical change at constant temperature and pressure is enthalpy of reaction (H), and it’s a thermodynamic quantity, q=ΔH [8].
Molecules, ions, or atoms containing at least an unpaired electron in the valance shell are called free radicals. Free radicals are chemically reactive, unstable, and mostly short lived. Heat, electrolysis, electrical discharge, and ionizing radiation can generate free radicals. Free radicals are intermediates in many chemical reactions. Free radicals are important in atmospheric chemistry, combustion, plasma chemistry, polymerization, biochemistry, and they are important in many chemical reactions [9].
In 2016 Hang Wang studied the thermodynamic properties of fluorinated methanol using CBS-QB3, M06-2X, WB97X, W1U, M06, B3LYP, CBS-APNO and G4 Calculations. Small standard deviation suggests good error cancellation of work reactions and accuracy. Small values for standard deviations was obtained in calculations using M06-2x/6-31+g (d,p) Gaussian method; it is an accurate method to calculate Enthalpy of fluorinated alcohols, it shows the second smallest standard deviation after CBS-QB3 method of calculation. Enthalpies of fluorinated methanol were studies in the past [10].
Halogenated compounds have low reactivity, are highly stable, and are used in industry. They are of concern to the environment due to their persistence in the environment, and its widespread use. Their thermochemical properties must be studied in order to understand the reduction and oxidation reactions involving these molecules [11].
Computational method
The global hybrid meta-GGA density functional approximation, GGA, DFT Gaussian M-062x/6-31+g (d,p) method of calculation has been used to initially analyze frequencies, optimized structures, and thermo energies of the molecules studied. In the GGA, generalized gradient approximation, the density functional depends on the down and up spin densities and the reduced gradient. In the meta GGA, the functional also depends on the up and down spin kinetic energy densities. A hybrid GGA is a combination of GGA with Hartree Fock exchange. The hybrid meta GGA is a combination of meta GGA with Hartree-Fock exchange [12].
A series of Isodesmic Reactions and composite calculations were employed to calculate enthalpy of formation of tri-, terta-, and penta-fluorinated ethanols. Calculations were performed using Gaussian 16 program. All reported calculations of enthalpy of formation are for standard conditions of 1 atm pressure and 298K. The Gaussian M-062 x/6-31+g (d,p) level of calculation has been used for this study as this method of calculation were successfully employed in the past when applied to fluoro hydrocarbons 6 with small reported standard deviations values [13] .
The standard deviation is calculated using the following formula [14].
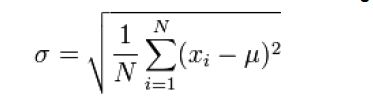
Where, Xi is the mean; the average of the numbers, μ is the actual numbers to be calculated the standard deviation of, and
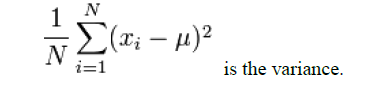
Isodesmic and isogyric reaction
The Gaussian M-062x/6-31+g (d,p) method of calculation has been used to calculate the enthalpy of formation of tri-, terta-, and penta fluorinated ethanol’s and its radicals. Work reactions and reference species have been used to calculate enthalpy of formation of fluorinated ethanol’s using this method.
To cancel any systematic error in the molecular orbital calculations using this method, the number of each type of bond must be conserved in each isodesmic reactions. By the careful choice of the isodesmic reactions, all enthalpies of formation calculations are allowed to accuracies close to experimental values.
The ΔHf298°K values of all reference species but the fluoroethanols are known, the ΔHf298°K of the target species fluoro ethanols, is obtained from this data and the calculated ΔHrxn, 298°. ΔHf298°K calculated using two different reference molecules are within ± (0 to 0.60 Kcal mol-1).
Reference species
Table 1 lists the standard enthalpy of formation for the reference species used in isodesmic reactions with their uncertainties. Table 2 provides all calculated standard enthalpy of formation values, ΔfHo (298) for of tri-, terta-, and penta fluorinated ethanol’s and its radicals [15-16].
| Species | ΔfHO (298) | Species | ΔfHO (298) |
|---|---|---|---|
| CH3F | -56.54 ± 0.07a -56.62 ± 0.48h |
CH3OOH CH3CH2OOH |
-30.96 ± 0.67b -38.94 ± 0.81b |
| CH3CH2F | -65.42 ± 1.11a | CH3CH2CH2OOH | -44.03 ± 0.67b |
| CH3CH2CH2F | -70.24 ± 1.30a | CH3OO• | 2.37 ± 1.24b |
| CH2F2 | -108.07 ± 1.46a -107.67 ± 0.48h |
CH3CH2OO• CH3CH2CH2OO• | -6.19 ± 0.92b -11.35 ± 1.24b |
| CH3CHF2 | -120.87 ± 1.62a | CH4 | -17.81 ± 0.01c |
| CH3CH2CHF2 | -125.82 ± 1.65a | CH3CH3 | -20.05 ± 0.04c |
| CHF3 | -166.71 ± 1.97h -166.09 ± 0.48h |
CH3CH2CH3 CH3CH2CH2CH3 | -25.01 ± 0.06i -30.07 ± 0.08i |
| CH3CF3 | -180.51 ± 2.05a | CH3O• | 5.15 ± 0.08 c |
| CH3CH2CF3 | -185.48 ± 2.15a | CH3CH2O• | -3.01d |
| CH3• | 34.98 ± 0.02c | OH | 8.96 ± 0.01c |
| CH3CH2• | 28.65 ± 0.07c | CH3OH | -47.97 ± 0.04c |
| CH3CH2CH2• | 24.21 ± 0.24gj 24.18i |
CH3CH2OH | -56.07 ± 0.05i |
| H | 52.10c | HOO• | 2.94cj |
| O | 59.57c | HOOH | -32.39 ± 0.04fj -32.37i |
| CF4 | -223.15 | CHF2CHF2 | -212.13 |
| CF3CH2CHF2 | -286.18 | CF3CHF2 | -267.79 |
Table 1. Reference Species in the Isodesmic reactions standard enthalpy of formation values (kcal mol-1) 15.
Standard enthalpy values
Isodesmic work reactions from M-062x/6-31+g (d,p) method of calculation has been utilized to perform calculation of standard enthalpy of formations for tri-, tertra-, and penta- fluorinated ethanol’s and some radicals. Reference species and their standard enthalpy of formation along with their uncertainties have been used in all isodesmic work reactions. The calculated sum of thermal enthalpies using the Gaussian M06-2x/6-31+g (d,p) Level of Theory for all target fluorinated ethanol’s, their radicals, and for reference species has also been used (Figure 1).

Figure 1: Calculated the Number of fluorine atoms per molecules.
The standard enthalpy of formation in kcal mol−1 for all reference species are listed in Table 1. Standard deviation values 14 are listed for all calculated Gaussian M06-2x/6-31+g (d,p) standard enthalpy of formations values. The calculated Gaussian M06-2x/6-31+g (d,p) standard enthalpy of formation for tri-, tertra-, and penta fluorinated ethanol’s and some radicals are listed Table 2.
| Isodesmic reactions target specie |
ΔH° Rxn (298) Hartbees | ΔH° Rxn (298) Kcal/mole1 | ΔfH° (298) kcal mol-1 |
Error kcal mol-1 |
|---|---|---|---|---|
| CF3CH2OH+CH4=CH3CH2OH+CHF3 -452.696144-40.447961-154.92666-338.204669 -17.81 -56.21 -166.71 |
0.01277 | 8.013303 | -213.1 | ± 2.03 |
| CF3CH2OH+CH3CH3=CH3CH2OH+CH3C -452.696144 -79.71776 -154.926666 -377.492814-20.05-56.21-180.51 |
-0.00557 | -3.49398 | -213.2 | ± 2.14 |
| Reported ΔfHo (298) kcal mol-1 | -213.15±2.09 | |||
| Standard deviation over rxns 0.05 kcal mol-1 | ||||
| CF3CH•OH+CH3CH3=CH3CH2O•+CH3CF3 -452.048269 -79.717768 -154.268107 -377.492814-20.05 -3.01 -180.51 |
0.005116 | 3.210341 | -166.7 | ± 2.09 |
| CF3CH•OH+CH4=CH3CH2O•+CHF3 -452.048269 -40.447961 -154.268107 -338.204669-17.81-3.01-166.71 |
0.023454 | 14.71762 | -166.6 | ± 1.98 |
| Reported ΔfHo (298) kcal mol-1 | -166.65±2.04 | |||
| Standard deviation over rxns 0.05 kcal mol-1 | ||||
| CF3CH2O•+CH3CH2CH3=CH3CH2O•+CH3CH2CF3 -452.023032-118.99092-154.268107-416.766483-25.02-3.01-185.48 |
-0.02064 | -12.9537 | -150.5 | ± 2.21 |
| CF3CH2O•+CH4=CH3CH2O•+CHF3 -452.023032-40.447961-154.268107-338.204669-17.81-3.01-166.71 |
-0.00178 | -1.11885 | -150.8 | ± 1.98 |
| Reported ΔfHo (298) kcal mol-1 | -150.70±2.09 | |||
| Standard deviation over rxns 0.16 kcal mol-1 | ||||
| CHF2CHFOH+CH3CH3=CH3CH2OH+CH3CF3 -452.686514-79.717768-154.926666-377.492814-20.05-56.21-180.51 |
-0.0152 | -9.5369 | -207.1 | ± 2.14 |
| CHF2CHFOH+CH4=CH3CH2OH+CHF3 -452.68651440.447961-154.926666-338.204669-17.81-56.21-166.71 |
0.00314 | 1.970381 | -207.1 | ± 2.03 |
| Reported ΔfHO(298) kcal mol-1 | -207.10 ± 2.09 | |||
| Standard deviation over rxns 0.00 kcal mol-1 | ||||
| C•F2CHFOH+CH4=CH3CH2O• +CHF3 -452.032761-40.447961-154.268107-338.204669-17.81-3.01-166.71 |
0.007946 | 4.986194 | -156.9 | ± 1.98 |
| C•F2CHFOH+CH3CH3= CH3CH2O•+CH3CF3 -452.032761-79.717768-154.268107-377.492814-20.05 -3.01 -180.51 |
-0.01039 | -6.52108 | -156.9 | ± 2.09 |
| Reported ΔefHo (298) kcal mol-1 | -156.90 ± 2.04 | |||
| Standard deviation over rxns 0.00 kcal mol-1 | ||||
| CHF2C•FOH+CH3CH3=CH3CH2O•+CH3CF3 -452.028711-79.717768-154.268107-377.492814-20.05-5.01-180.51 |
-0.01444 | -9.0625 | -154.4 | ± 2,09 |
| CHF2C•FOH+CH3CH2CH3=CH3CH2O•+CH3CH2CF3 -452.028711-118.990915-154.268107-416.766483-25.02-5.01-185.48 |
-0.01496 | -9.39006 | -154.1 | ± 2.21 |
| Reported ΔfHO (298) kcal mol-1 | -154.30 ± 2.15 | |||
| Standard deviation over rxns 0.16 kcal mol-1 | ||||
| CHF2CHFO•+CH4=CH3CH2O•+CHF3 -452.019492-40.447961-154.268107-338.20466-17.81 -3.01-166.71 |
-0.00532 | -3.34024 | -148.6 | ± 1.98 |
| CHF2CHFO•+CH3CH2CH3=CH3CH2O• + CH3CH2CF3 -452.019492-118.99092-154.268107-416.766483-25.02 -3.01-185.48 |
-0.02418 | -15.1751 | -148.3 | ± 2.21 |
| Reported ΔfHo (298) kcal mol-1 | -148.50 ± 2.09 | |||
| Standard deviation over rxns 0.16 kcal mol-1 | ||||
| CH2FCF2OH+CH4=CH3CH2OH+CHF3 -452.696009-40.447961-154.926666-338.204669-17.81 -56.21-166.71 |
0.012635 | 7.928589 | -213.0 | ± 2.03 |
| CH2FCF2OH +CH3CH3=CH3CH2OH+CH3CF3 -452.696009-79.717768-154.926666-377.492814-20.05 -56.21-180.51 |
-0.0057 | -3.57869 | -213.1 | ± 2.14 |
| Reported ΔfHo (298) kcal mol-1 | -213.05 ± 2.09 | |||
| Standard deviation over rxns 0.05 kcal mol-1 | ||||
| C•HFCF2OH+CH4=CH3CH2O•+CHF3-452.03698640.447961-154.268107-338.204669-17.81-3.01-166.71 | 0.012171 | 7.637424 | -159.5 | ± 1.98 |
| C•HFCF2OH+CH3CH3=CH3CH2O•+CH3CF3 -452.036986-79.717768-154.268107-377.492814-20.05-3.01-180.51 |
-0.00617 | -3.86985 | -159.6 | ± 2.09 |
| Reported ΔfHo (298) kcal mol-1 | -159.55 ± 2.04 | |||
| Standard deviation over rxns 0.08 kcal mol-1 | ||||
| CH2FCF2O•+CH4=CH3CH2O•+CHF3 -452.019556-40.447961-154.268107-338.204669 -17.81-3.01-166.71 |
-0.00526 | -3.30008 | -148.6 | ± 1.98 |
| CH2FCF2O•+CH3CH3=CH3CH2O•+CH3CF3 -452.019556-79.717768-154.268107-377.492814-20.05 -3.01-180.51 |
-0.0236 | -14.8074 | -148.7 | ± 2.09 |
| Reported ΔfHo (298) kcal mol-1 | -148.65 ± 2.04 | |||
| Standard deviation over rxns 0.05 kcal mol-1 | ||||
| CF3CFHOH+CH4=CH3CH2OH+CF4 -551.950082-40.447961-154.926666-437.46666-17.81 -56.21-223.15 |
0.004717 | 2.959965 | -264.5 | ± 0.10 |
| CF3CFHOH+CH3CH3=CH3CH2OH+CHF2CHF2 -551.950082-79.717768-154.926666-476.71600-20.05-56.21-212.13 |
0.025183 | 15.80258 | -264.1 | ± 0.09 |
| Reported ΔfHo (298) kcal mol-1 | -264.30 ± 0.10 | |||
| Standard deviation over rxns 0.20 kcal mol-1 | ||||
| CF3C•FOH+ CH4=CH3CH2O•+CF4 -551.293882-40.447961-154.268107-437.4666-17.81-3.01 -223.15 |
0.007076 | 4.440261 | -212.8 | ± 0.10 |
| CF3C•FOH+CH3CH3=CH3CH2O•+CHF2CHF2 -551.293882-79.717768-154.268107-476.716001-20.05-3.01 -212.13 |
0.027542 | 17.28288 | -212.4 | ± 0.40 |
| Reported ΔfHo (298) kcal mol-1 | -212.60 ± 0.30 | |||
| Standard deviation over rxns 0.20 kcal mol-1 | ||||
| CF3CFHO•+CH4 =CH3CH2O•+CF4 -551.281264-40.447961-154.268107-437.46666-17.81 -3.01-223.15 |
-0.00554 | -3.47766 | -204.9 | ± 0.10 |
| CF3CFHO•+CH3CH3=CH3CH2O•+CHF2CHF2 -551.281264-79.717768-154.268107-476.716001-20.05 -3.01-212.13 |
0.014924 | 9.364959 | -204.5 | ± 0.40 |
| Reported ΔfHo (298) kcal mol-1 | -204.70 ± 0.30 | |||
| Standard deviation over rxns 0.20 kcal mol-1 | ||||
| CF2HCF2OH+CH4=CH3CH2OH+CF4 -551.947553-40.447961-154.926666-437.46666-17.81-56.21-223.15 |
0.002188 | 1.372992 | -262.9 | ± 0.10 |
| CF2HCF2OH+CH3CH3=CH3CH2OH+CHF2CHF2 -551.947553 -79.717768-154.926666-476.716001-20.05 -56.21-212.13 |
0.022654 | 14.21561 | -262.5 | ± 0.10 |
| Reported ΔfHo (298) kcal mol-1 | -262.70 ± 0.10 | |||
| Standard deviation over rxns 0.20 kcal mol-1 | ||||
| C•F2CF2OH+CH4=CH3CH2O•+CF4 -551.286788 -40.447961-154.268107-437.46666-17.81 -3.01-223.15 |
-1.8E-05 | -0.0113 | -208.3 | ± 0.10 |
| C•F2CF2OH+CH3CH3=CH3CH2O•+CHF2CHF2 -551.286788-79.717768-154.268107-476.716001-20.05-3.01-212.13 |
0.020448 | 12.83132 | -207.9 | ± 0.40 |
| Reported ΔfHo(298) kcal mol-1 | -208.10 ± 0.30 | |||
| Standard deviation over rxns 0.20 kcal mol-1 | ||||
| CF2HCF2O•+CH4=CH3CH2O•+CF4 -551.26873-40.447961-154.268107-437.46666-17.81-3.01 -223.15 |
-0.01808 | -11.3429 | -197.0 | ± 0.10 |
| CF2HCF2O• + CH3CH3=CH3CH2O•+CHF2CHF2 -551.26873-79.717768-154.268107-476.716001-20.05-3.01-212.13 |
0.00239 | 1.499749 | -196.6 | ± 0.40 |
| Reported ΔfHo(298) kcal mol-1 | -196.80 ± 0.30 | |||
| Standard deviation over rxns 0.20 kcal mol-1 | ||||
| CF3CF2OH+CH3CH2CH3=CH3CH2OH+CF3CH2CHF2 -651.207522-118.99092-154.926666-615.260935-25.02-56.21-286.18 |
0.010836 | 6.799698 | -324.2 | ± 0.10 |
| CF3CF2OH+CH3CH3=CH3CH2OH+CF3CHF2 -651.207522 -79.717768 -154.926666 -575.976217-20.05 -56.21-267.79 |
0.022407 | 14.06062 | -318.0 | ± 1.70 |
| CF3CF2OH + CH4 =CF3OH+CH3CHF2 -651.207522-40.447961-413.437575-278.225303-17.81-218.11-12087 |
-0.00739 | -4.64044 | -316.5 | ± 1.70 |
| Reported ΔfHo (298) kcal mol-1 | -319.60 ± 1.20 | |||
| Standard deviation over rxns 3.30 kcal mol-1 | ||||
| CF3CF2O•+CH4=CHF2O•+CH3CF3 -650.527081-40.447961-313.498636-377.492814-17.81 -97.82-180.51 |
-0.01641 | -10.2962 | -250.2 | ± 2.06 |
| CF3CF2O•+CH3CH2CH3=CH3CH2O•+CF3CH2CHF2 -650.527081-118.990915-154.268107-615.260935-25.02 -3.01-286.18 |
-0.01105 | -6.93148 | -257.2 | ± 0.10 |
| CF3CF2O•+CH3CH3=CH3CH2O•+CF3CHF2 -650.527081-79.717768-154.268107-575.976217-20.05-3.01-267.79 |
0.000525 | 0.329443 | -251.1 | ± 1.70 |
| Reported ΔfHo(298) kcal mol-1 | -252.80 ± 1.30 | |||
| Standard deviation over rxns 3.11 kcal mol-1 | ||||
Hartbees, kcal mole-1
*SD Standard Deviation kcal mol-1
Errors reported avg of sum of uncertainties in rxn’s reference species
Table 2. Isodesmic reactions used in calculating standard enthalpy of formation, ΔH° Rxn for tri-, terta-, and penta-fluorinated ethanol’s and its radicals using the Gaussian M06-2x/6-31+g (d,p) level of theory.
Bond energies
The difference in bond dissociation energies of products and reactants for the hemolysis is called the bond dissociation energy. The bond dissociation energy values don’t depend on the pathway by which it occurs, it doesn’t depend on how bonds are formed or on how bonds break, therefore bond dissociation energies are state functions. Energetics of chemical processes can be assessed using bond dissociation energy values.
The calculated bond dissociation energy values in kcal/mol for Methyl C-H, Ethyl C-H, and hydroxyl O-H bonds are listed in Table 3.
| Reactions | Bond dissociation energya (Kcal mol-1) | *Error Kcal mol-1 |
|---|---|---|
| CF3CH-HOH CF3CH-HOH=H•+CF3CH•OH -213.15 ± 2.09 52.10-166.65 ± 2.04 CF3CH2O-H CF3CH2O-H=H• +CF3CH2O• -213.15 ± 2.09 52.10-150.70 ± 2.09 C-HF2CHFOH C-HF2CHFOH=H•+C•F2CHFOH -207.10 ± 2.09 52.10-156.90 ± 2.04 CHF2C-HFOH CHF2C-HFOH =H•+CHF2C•FOH -207.10 ± 2.09 52.10-154.30 ± 2.15 CHF2CHFO-H CHF2CHFO-H = H•+CHF2CHFO• -207.10 ± 2.09 52.10-148.50 ± 2.09 HC-HFCF2OH HC-HFCF2OH =H•+C•HFCF2OH -213.05 ± 2.09 52.10-159.55 ± 2.04 CH2FCF2O-H CH2FCF2O-H=H•+CH2FCF2O• -213.05 ± 2.09 52.10 -148.65 ± 2.04 CF3CF-HOH CF3CF-HOH=H•+CF3CF•OH -264.30 ± 0.10 52.10 -212.60 ± 0.30 CF3CFHO-H CF3CFHO-H=H•+CF3CFHO• -264.30 ± 0.10 52.10-204.70 ± 0.30 CF2-HCF2OH CF2-HCF2OH=H•+CF2• CF2OH -262.70 ± 0.10 52.10-208.10 ± 0.30 CF2HCF2O-H CF2HCF2O-H=H•+CF2HCF2O• -262.70 ± 0.10 52.10-196.80 ± 0.30 CF3CF2O-H CF3CF2O-H=H•+CF3CF2O• -319.60 ± 1.20 52.10-252.80 ± 1.30 |
98.60 ± 2.07
102.30 ± 2.07 104.90 ± 2.10
103.80 ± 0.20
106.70 ± 0.20 118.00 ± 0.20
118.90 ± 1.30
|
± 2.07 ± 2.09 ± 2.07 ± 2.10 ± 2.09 ± 2.07
± 0.20
± 0.20
± 0.20 ± 1.30 |
*Errors reported avg of sum of uncertainties in rxn’s reference specie
Table 3. Bond Dissociation Energy (BDE’s) of Tri-, Trtra-, and Penta Fluorinated Ethanol’s using Gaussian M-062x/6-31+g (d,p) method of calculation.
Optimized structures
The Gauss view software has been used along with gaussian output files to provide a picture of the Gaussian M06-2x/6-31+g (d,p) optimized structure for each molecule in this study (Table 4).
CF3CH2OH 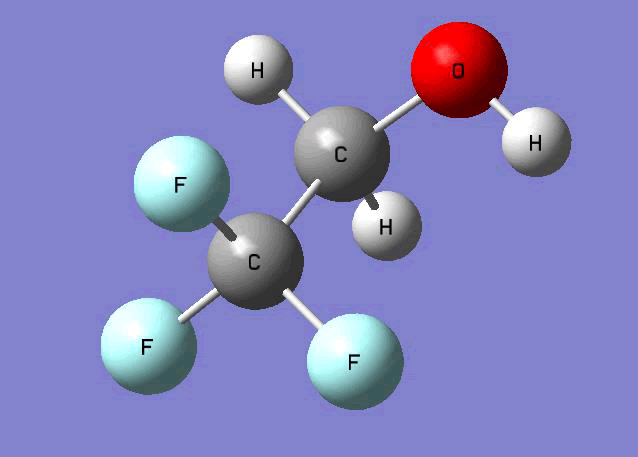 |
CF3CH•OH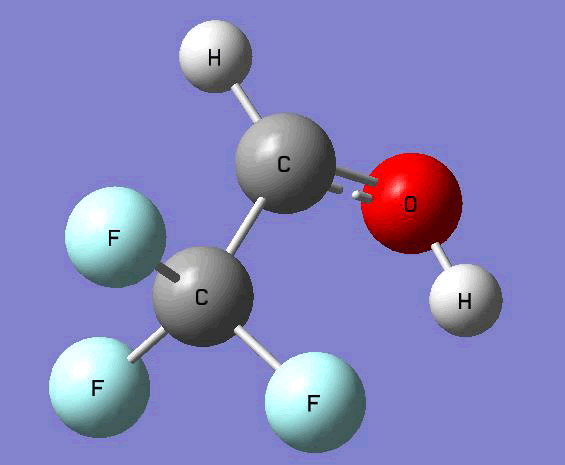 |
CF3CH2O•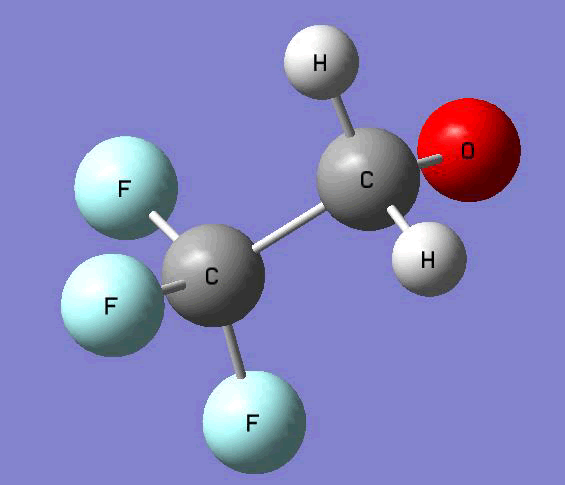 |
CHF2CHFOH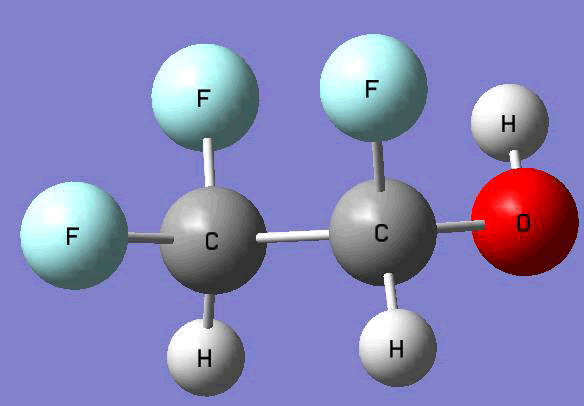 |
C•F2CHFOH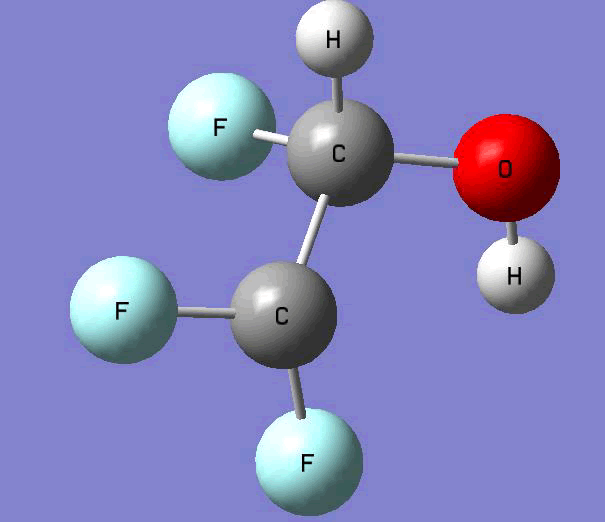 |
CHF2C•FOH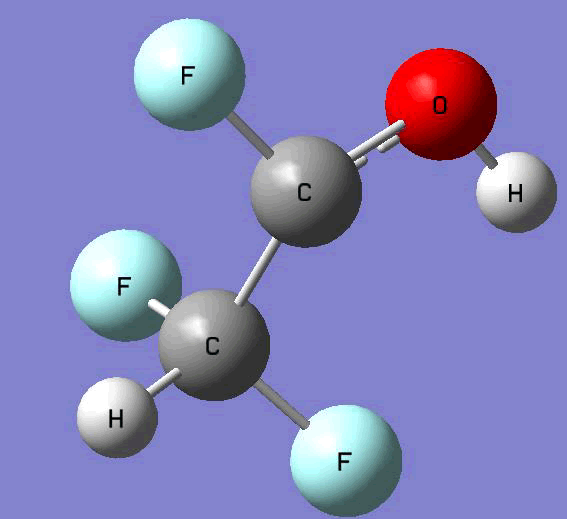 |
CHF2CHFO• 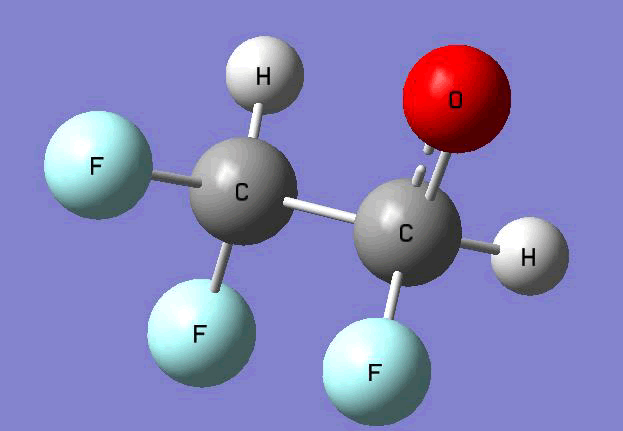 |
CH2FCF2OH 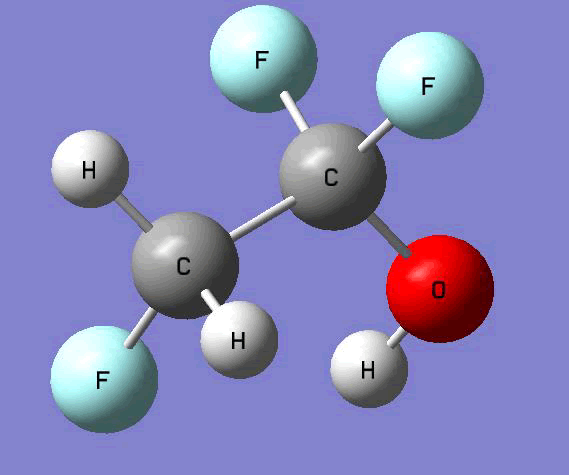 |
C•HFCF2OH 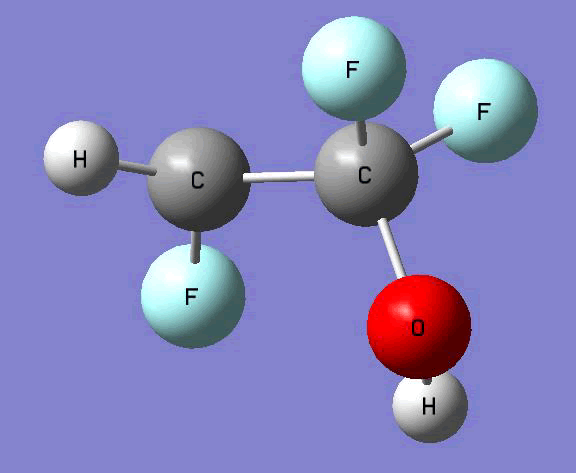 |
CH2FCF2O• 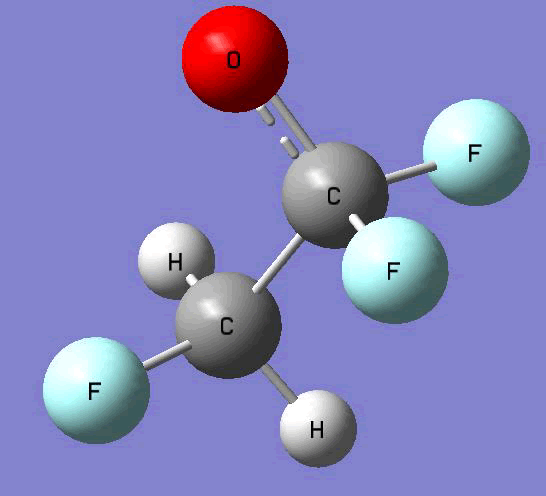 |
CF3CFHOH 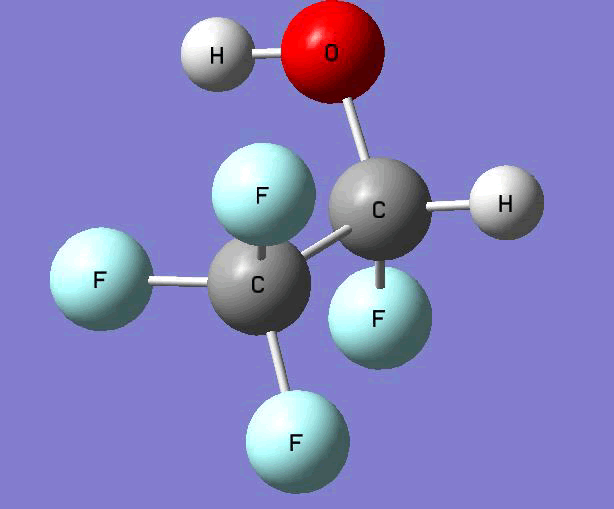 |
CF3CF•OH 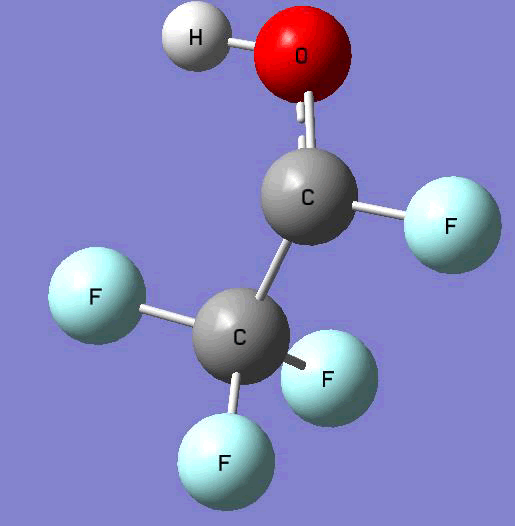 |
CF3CFHO• 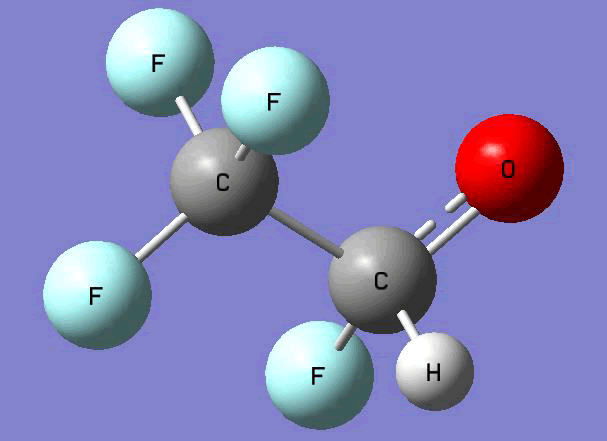 |
CF2HCF2OH 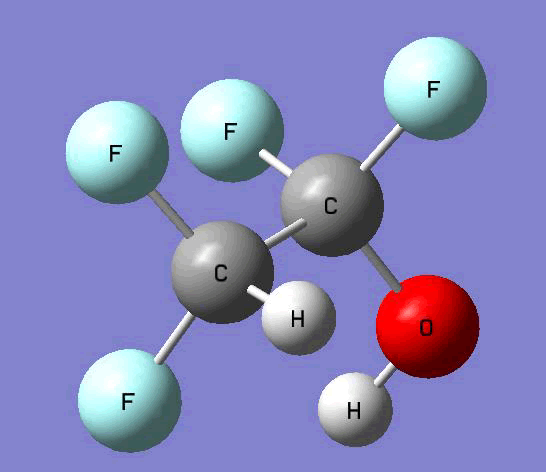 |
C•F2CF2OH 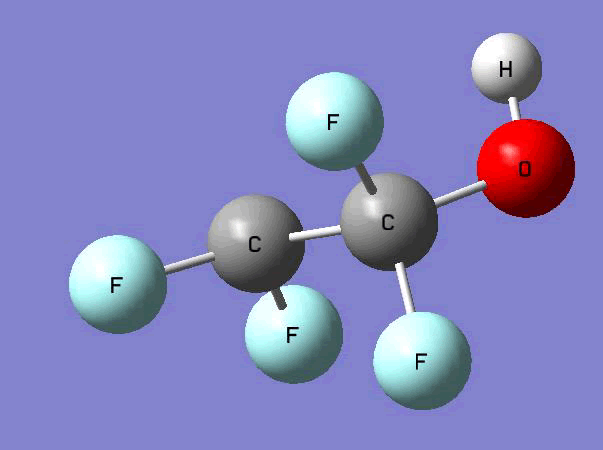 |
CF2HCF2O• 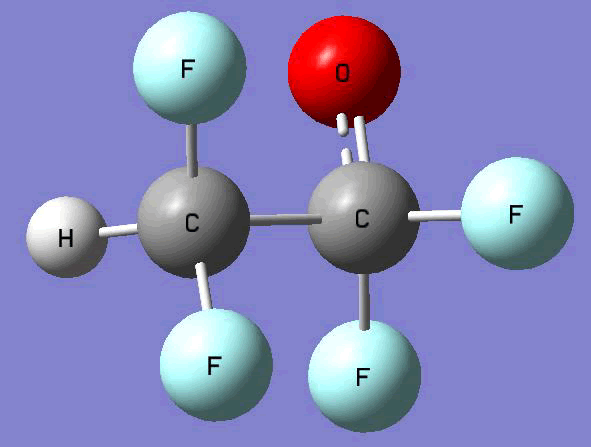 |
CF3CF2OH 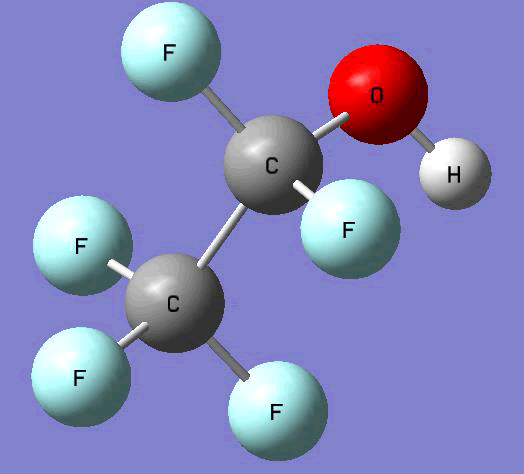 |
CF3CF2O• 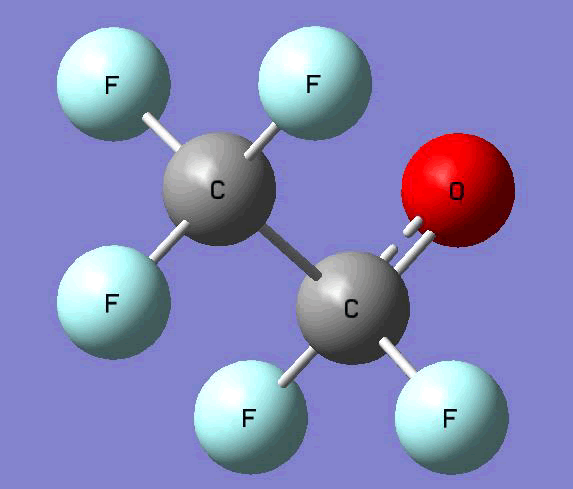 |
Table 4. Optimized geometries for target tri-, tetra-, and penta- fluorinated ethanol’s and their related radicals calculated by Gaussian M06-2x/6-31+g (d,p) level of theory.
Cartesian coordinates
The cartesian coordinates for target tri-, tetra-, and penta- fluorinated ethanol’s and their related radical’s at the Gaussian m062x/6-31+g (d,p) level of theory are listed in Table 5.
| Molecule | Cartesian coordinates |
| CF3CH2OH | 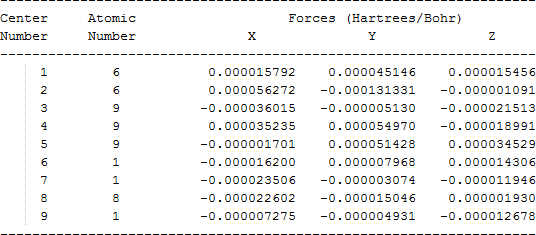 |
| CF3C•HOH | 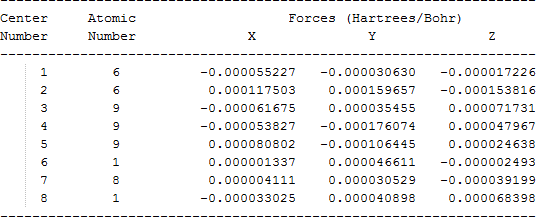 |
| CF3CH2O• | 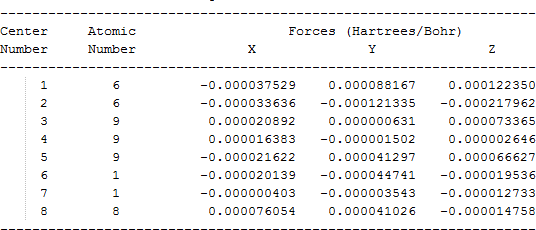 |
| CHF2CHFOH | 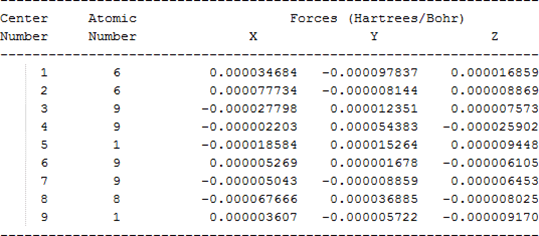 |
| C•F2CHFOH | 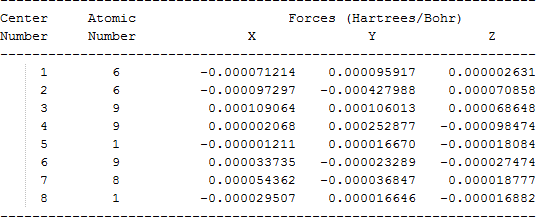 |
| CHF2C•FOH | 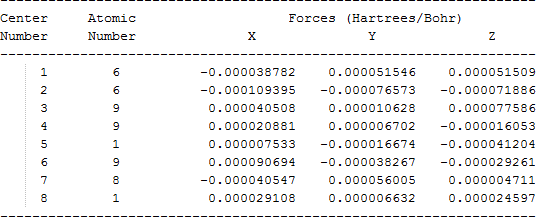 |
| CHF2CHFO• | 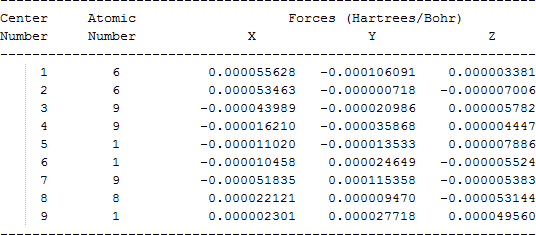 |
| CH2FCF2OH | 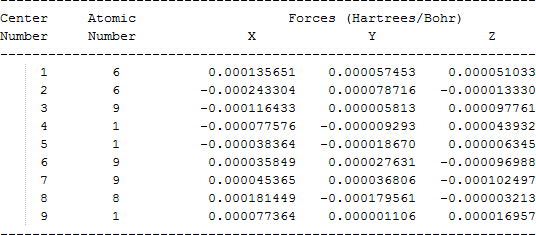 |
| C•HFCF2OH | 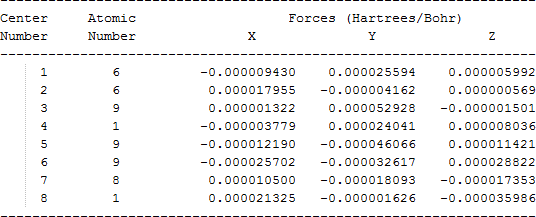 |
| CH2FCF2O• | 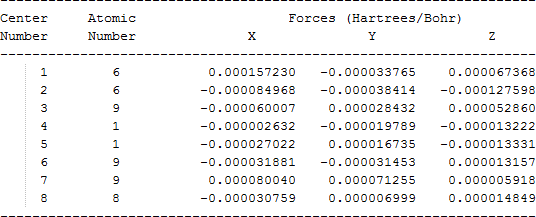 |
| CF3CFHOH | 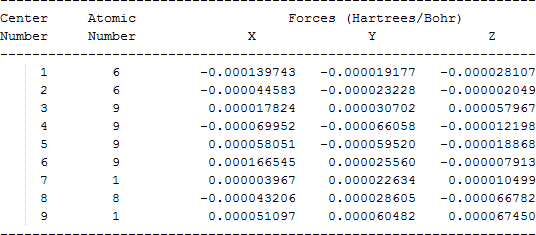 |
| CF3C•FOH | 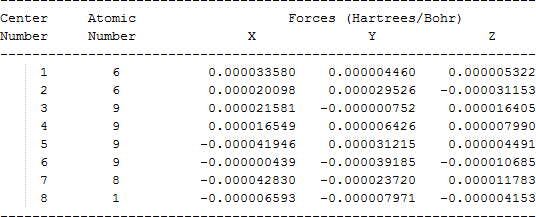 |
| CF3CFHO• | 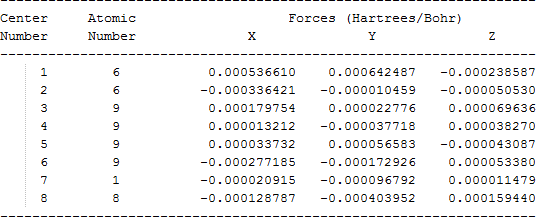 |
| CF2HCF2OH | 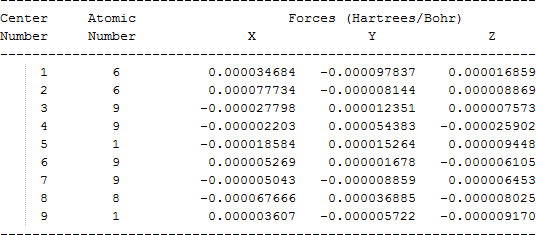 |
| C•F2CF2OH | 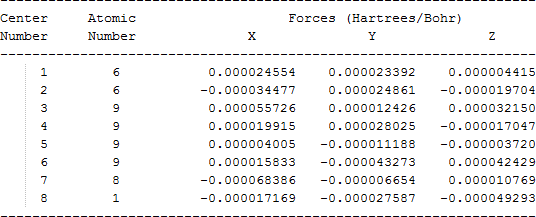 |
| CF2HCF2O• | 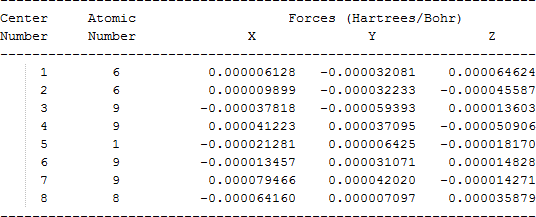 |
| CF3CF2OH | 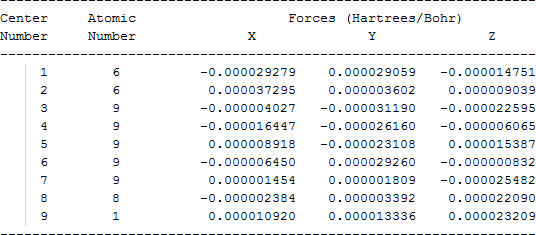 |
| CF3CF2O• | 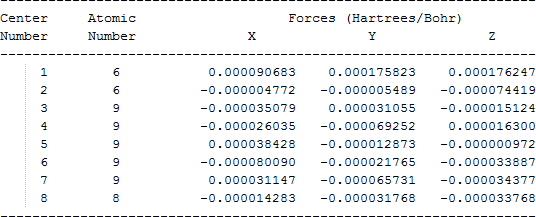 |
Table 5. Cartesian coordinates in angstroms for target fluorinated ethanol and their related radical’s geometries at the m062x/6-31+g (d,p) level of theory.
We acknowledge the NJIT advanced research computing services for significant help in providing the computer calculation software.
The ab initio and Global-hybrid meta-GGA density function method, Gaussian M06-2x/6-31+g (d,p) has been used to calculate thermodynamic properties of 18 tri-, tetra-, and penta- fluorinated ethanol’s and their related radicals. Standard enthalpy of formation at the Gaussian M06-2x/6-31+g (d,p) calculation level has been calculated using multiple work reactions, work reactions were employed for cancellation of calculation errors.
The thermochemical properties: the O—H, secondary methyl C—H, and ethyl C-H Bond Dissociation Energies (BDEs), and standard enthalpy of formation (298 K) values has been calculated for tri-, tetra-, and penta- fluorinated Ethanol’s and their related radicals: CH3CH2-xFxOH, CH3-xFxCH2-xFxOH, and CH2-xFxCH2OH
The C-H bond dissociation energies range from 98.6 to 104.9 Kcal mol-1 on the secondary ethyl carbons, and from 103.3 to 106.7 Kcal mol-1 on the primary methyl carbons. The O-H bond energies range from 110.7 to 118.9 Kcal mol-1.
Calculated values for the O-H bond energies for penta fluorinated ethanol are higher than those of O-H bond energies for tri-fluorinated ethanol. The calculated the O-H bond energies increased by intruding more fluorine atoms to either methyl or ethyl carbons.
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]
[Crossref] [Googlescholar] [Indexed]