Research & Reviews: Journal of Chemistry
e-ISSN: 2319-9849
e-ISSN: 2319-9849
Department of Chemistry, Faculty of Science, Benghazi University, Benghazi, Libya
Received: 06/11/2013; Revised: 07/12/2013; Accepted: 25/11/2013
Visit for more related articles at Research & Reviews: Journal of Chemistry
The compounds which were synthesized and presented in this project serve as stable models of reactive intermediates, proposed in catalytic transformations with the ruthenium counterparts. It is also of general interest to study the reactivity of trimethylphosphine stabilized iron compounds toward aromatic imines and ketones. Other than some cyclomanganated products, there are no examples of cyclometalated products of 3d row transition metals with imines. In this section, a review of reactions in the activation and functionalization of C-H bonds by solution-phase transition metal-based systems are presented, with an emphasis on the activation of aromatic C-H bonds. Phenyl ketimines react smoothly under mild conditions with low- valent iron (0) adducts to form five-membered metallacycles. The reaction of Diphenylketimine and tert-butylphenylketimine with Fe(PMe3)4 generates a hydrido-iron (II) complex 1,2 respectively. Iodomethane reactions of hydrido-iron compound form iodo-iron (II) complex 3. Benzylic imine react with Fe(CH3)2(PMe3)4 by elimination of methane to afford hexacoordinate methyl-iron(II) complex 4. All complexes are given from 1- 4 were characterized through IR, NMR and X-ray diffraction was discussed.
Crystal structures; Iron complexes; Imine reactions
Organometallic chemistry lies at the interface between classical organic and inorganic chemistry because it looks at the interaction between inorganic metal ions and organic molecules. The field has provided some powerful new synthetic methods in organic chemistry and is beginning to make links with biochemistry with the discovery of several metallo- enzymes that involve organometallic intermediates [1-4]. Organometallic ideas have been useful in interpreting the chemistry of metal surfaces and of metal colloids. The controlled pyrolysis of organometallic species has proved to be a useful way of preparing solid-state material with unusual properties [5,6]. Public concern for the environment has led to the rise of ?green chemistry?, the purpose of which is to minimize the production of chemical waste in industry and commerce. One way to do this is to use catalysts rather than stoichiometric reagents to bring about reactions. Many commercially important processes that rely on transition metal organometallic complexes as catalysts have been developed, and such applications are likely to gain more importance in the future [7]. The carbon – hydrogen bond of alkanes cannot usually be regarded as functional group. Its unique position in organic chemistry is well illustrated by the standard representation of organic molecules: the presence of C-H bonds is indicated simply by the absence of any other bond. This ?invisibility? of C-H bonds reflects both their ubiquitous nature and their lack of reactivity. With these characteristics in mind it is clear that if the ability to selectivity functionalize C-H bonds were well developed, it could potentially constitute the most broadly applicable and powerful class of transformations in organic synthesis. Realization of such potential could revolutionize the synthesis of organic molecules ranging in complexity from methanol to the most elaborate natural or unnatural products. The activation of C-H bonds by ?organometallic routes? (i.e., those involving the formation of a bond between carbon and a metal center) is a very large, diverse and highly active field [8-14]. The first reported of ?C-H activation? by a transition metal complex is often attributed to Chatt. [15] C-H bond activations are often classified as proceeding via either nucleophilic (oxidative addition) or electrophilic modes. However, these two classes of C-H activation have much in common, perhaps even more so than the corresponding modes of H2 activation. Both reactions appear to proceed through a σ-bond intermediate [16-18]. Even more striking, putatively electrophilic activations, in most cases, proceed via complete oxidative addition (followed by deprotonation of the resulting metal hydride) [19].
All syntheses and manipulations were carried out under an inert atmosphere of argon or nitrogen by using modified Schlenk techniques [20]. This type of apparatus offers an opportunity of carrying out a complete synthesis in a closed system, and in one run. Synthesis, transport and storage of chemicals were done under an atmosphere of purified Argon (BTS catalyst) [21]. Solvents (THF, diethyl ether, pentane) were dried according to known procedures and were freshly distilled prior to use. All reagents (Aldrich, Acros, Fluka, or Lancaster) were used as purchased without further purification.
Trimethylphosphine PMe3[22], Fe(PMe3)4[23], FeMe2(PMe3)4[24], (tert- Butylphenylketimine and Diphenylketimine [25,26], and were synthesized according to literature procedures. C, H, P, N analyses of air sensitive solids were carried by H. Kolbe micro-analytical laboratory, Mulheim /Ruhr. Infrared spectra (4000-400 cm-1), as obtained from Nujol mulls between KBr discs, were recorded on a Bruker FRA106 spectrometer. 1H, 13C and 31P NMR spectra were obtained from Bruker AVANCE 500, ARX 300 and AM 200 spectrometers. 13C and 31PNMR resonances were obtained with broad-band proton decoupling. Assignment of 13C signals was supported by DEPT trace. Melting points were measured in capillaries sealed under argon atmosphere.
tert- ButylphenylketimineTo a stirred THF solution of 5.2 g (50.4 mmol) of benzonitrile and 31.5 ml (50.4 mmol, 1.6 M in THF) of tert.-butyl magnesium chloride, 250 mg (1.31 mmol) of copper (I) iodide at -78°C were added. The reaction mixture turned dark brown immediately. Progress of the reaction was monitored by thin layer chromatography. After 48 h, the reaction was completed, and the mixture was then filtered off from magnesium chloride followed by evaporation of the solvent in vacuo. The residue was distilled under reduced pressure to afford 4.2 g of an oily liquid (52%, boiling at 84°C). 1H NMR (300 MHz, [D8]THF, 297 K): δ = 1.22(s, 9H,C (CH3)3);7.18 – 7.24 (m, 2H, Ar- H); 7.32-736 (m, 3H, Ar-H); 9.43. (s, CH); 128.1 (s, CH); 142.9 (s, C); 180.4 (s, NC).
Hydrido[2-[(imino-KN)phenylmethyl]phenyl-KC]tris(trimethylphosphine)iron(II)(1)Diphenylketimine (0, 72 g, 3.99 mmol) in 20 ml of pentane were combined with Fe(PMe3)4 (1.44 g, 3.99 mmol) at -27°C to afford violet crystals (1). Yield 1.451g(78%); M.P 105 – 107 °C. (dec.). IR (Nujol) ν 3281 w (ν H-N=C); 1730vs ( ν Fe-H). 1H NMR (500 MHz, [D8]THF, 300 K): δ = – 16.4 ( dt, 2JP,H = 22.8 Hz, 2JP,H = 81.3 Hz, 1H, Fe-H); 0.94 ( s, 18 H, PCH3);1.42( d, 2JP,H = 4.7 Hz, 9H, PCH3); 6.72 ( t, 3JH,H = 7.0 Hz, 1H, Ar- H); 6.82 ( t, 3JH,H =7.1 Hz, 1H, Ar- H); 7.38 ( t, 3JH,H = 7.2 Hz, 2H, Ar- H); 7.47 ( t, 3JH,H = 7.8 Hz, 3H, Ar- H); 7.89 ( d, 3JH,H = 6.4 Hz, 1H, Ar- H); 9.40( s, 1H, N-H). 31P NMR (202 MHz, [D8]THF, 296 K): δ = 18.2 ( d, 2JP,P = 37.4 Hz, 2P, PCH3), 22.5 (t, 2JP,P = 37.4 Hz, 1P, PCH3). C22H38FeNP3 (465.29): Calc.: C, 56.79; H, 8.23; N 3.01; P 19.97 Found: C, 57.06; H, 7.58; N 2.96; P 20.09 %.
Hydrido[2-[(imino-KN)phenylmethyl]-1,1-dimethylethyl-KC]tris(trimethylphosphine)iron(II) (2)1.27g ( 3.53 mmol) of Fe(PMe3)4 in pentane were combined with 0.57g (3.53 mmol) of tert-butylphenylketimine to afford reddish violet crystals (2) at 4°C. Yield 0.753g (48%); M.P 118 - 120°C. (dec.). IR (Nujol) ν 3335 m (ν H-N=C); 3060 m, 3026 m (ν H-C=C); 1795 m (ν Fe-H); 1565 m (ν C=C); 1433 s ( δas PCH3); 1389 s; 1357 m; 1293 m, 1270 s (δs PCH3); 770 w, 730 s (γ C-Harom); 940 vs ( ρ1PCH3).1H NMR (500 MHz, [D8] THF, 300 K): δ = – 17.5( dt, 2JP,H = 21.8 Hz, 2JP,H= 80.0 Hz, 1 H, Fe-H ); 0.88( s, 18 H;PCH3), 1.40 (d, 2JP,H = 4.5 Hz, 9H, PCH3); 1.46 ( s, 9H; C(CH3)3); 6.71 (dd, 3JH,H = 6.5 Hz, 3JH,H = 7.6 Hz, 2H, Ar-H); 7.74 ( d, 3JH,H = 7.4 Hz, 1H, Ar-H); 7.85 ( d, 3JH,H = 6.5 Hz, 1H; Ar-H); 9.13 ( s, 1H, N-H). 13C NMR ( 125 MHz, [D8] THF, 300 K): δ = 22.3 ( tt, 3JP,C = 2.6 Hz, 1JP,C = 12.8 Hz, PCH3); 23.3 (td, 1JP,C = 12.8 Hz, 3JP,C = 2.6 Hz, PCH3); 29.8 ( s, C(CH3)3); 40.1 ( s, C(CH3)3); 117.6 ( s, CH); 186.1 ( m, C=N), 200.1 ( m, Fe-C). 31P NMR (202 MHz, [D8] THF, 300 K): δ 18.2 (d, 2JP, P = 36.5 Hz, 2P, PCH3); 23.7 (t, 2JP, P = 36.5 Hz, 1P, PCH3). C20H42FeNP3 (445.31): Calc.: C, 53.94; H, 9.51; N 3.15; P 20.87 Found: C, 53.89; H, 9.35; N 3.61; P 19.03 %
Iodo [2-[imino-kN) phenyl methyl]-2-methylphenyl-kC] tris-(trimethylphosphine) iron (II) (3)A sample containing 0.598g (1.285mmol) of compound (1) were combined with CH3I 0.227 g (1.00 ml, 1.285 mmol) in 50 mL of THF after 18 h formed a pink solution. The volatiles were removed in vacuo, and the resulting solid was extracted with pentane. Crystallization at -27°C afforded dark pink cubes: yield 0.635 g of 3 (77%); M.P < 143°C. (dec.). IR (Nujol) ν 3320 w ( ν H-N=C); 3070 vw, 1565 m ( ν H-C=C ); 1556 w ( ν C=C ); 1525 vw (ν C=N); 941 vs ( ρ1 PCH3); 738 s ( γ C-Harom); 716 s,( νasPC3 ); 660 m ( νs PC3 ). 1H NMR (500 MHz, [D8] THF, 300 K): δ = 1.06 ( s, 18H, PCH3), 1.61 ( d, 2JP,H = 5.9 Hz, PCH3); 6.69 ( m, 2H, Ar-H); 7.51 (d, 4JP,H = 7.4 Hz, 1H, Ar-H); 7.76 (d, 3JH,H = 6.7 Hz, 1H, Ar-H); 9.50 ( s, 1H, N-H). 31P NMR (202 MHz, [D8] THF, 300 K): δ 13.16 ( d, 2JP,P = 55.9 Hz, 2P, PCH3); 19.9 ( t, 2JP,P = 55.9 Hz, 1P, PCH3). C22H37FeINP3 (591.19): Calc.: C, 44.69; H, 6.31; N 2.37; P 15.72 Found: C, 44.39; H, 5.95; N 2.21; P 15.03 %.
Methyl [2-[(methyl imino-KN) methylphenyl-kC] tris-(trimethylphosphine)] iron (II) (4)1.0 g (2.56 mmol) of Fe(CH3)2(PMe3)4 in THF (20 ml) were combined with 0.31 g (2.56 mmol) of Nmethylbenzylidenimine in diethyl ether (50 ml) at -80°C. The mixture was warmed to 20°C and stirred for 18 h. During this period the solution turned red violet. The volatiles were removed in vacuo and the residue was extracted with pentane. Crystallization at 4 °C, afforded violet crystals (4) Yield 753 mg (65%); M.P. 115 – 117 °C (dec.). IR (Nujol): ν = 3053 vw, 3034 vw (ν H-C=C); 1572 m ( ν C=C); 1521 m (ν C=N); 1416 w (δas PCH3); 1294 m, 1276 s (δs PCH3); 1221 w; 1180 w (δs Fe-CH3); 1118 w; 1020 w; 996 w; 937 vs (ρ1 PCH3); 844 m (ρ2 PCH3);746 w, 730 m (γ C-Harom); 712 vw, 702 w (νas PC3); 677 w, 658 w(νs PC3). 1H NMR (500 MHz, [D8] THF, 298 K): δ = - 1.38 (s, 3H, Fe-CH3); 071(s, 18H, PCH3); 1.49 (s, 9H, PCH3); 3.65 (s, 3H, N-CH3); 6.74 (s, 1H, Ar-H); 6.85(s, 1H, Ar-H); 7.30 (s, 1H, Ar-H); 787(s, 1H, Ar-H); 8.48 (s, 1H, N=C-H). C18H38FeNP3 (417.25): Calc.: C, 51.81; H, 9.18; N 3.36; P 22.27 Found: C, 51.52; H, 9.38; N 3.37; P 22.27 %.
Reacting pentane solutions of Fe (PMe3)4 with diphenyl – and tert-butylphenylketimine (Eq.(1)) gives from which upon cooling dark violet prisms of crystal 1 and 2. The yields are 78% for 1 and 48% for 2 respectively. Compound 2 is moderately sensitive while compound 1 is extremely hygroscopic. Less than 1 bar argon decomposes between 105 – 118 ºC.
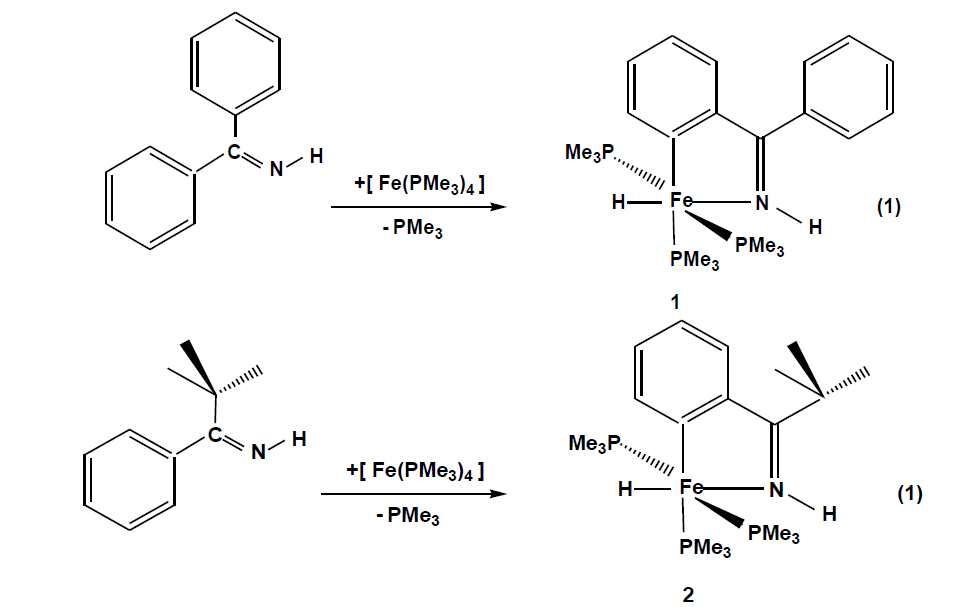
The FeH function is recognized in the infrared spectra by conspicuous ν(FeH) bands (1: 1730 cm-1, 2:1795cm-1)and ν(N-H) bands are observed with hypsochromic shifts between 45-70cm-1 indicating coordination through the N atom. Both spectra show all characteristic bands of coordinated PMe3 and the coordinated aromatic backbone, which in the 1H NMR spectra, Fe-H resonance appear at -16.4 for 1 and -17.5 for 2 as doublets of triplets due to the coupling of hydride nucleus with trans and cis disposed trimethylphosphine. Both compounds show two sets of enviromentally different PMe3 groups which indicate that the iron centers have an octahedral coordination. The 31P NMR spectra of the two compounds, which are temperature invariant, show coupling of 37 Hz for two sets of PMe3 groups. The PMe3 group trans to the metalated carbon gives a triplet with a resonance at around 23 ppm and the two isochronic trans phosphines appears at 18 ppm as doublets in both cases. The molecular structure of 1 (Fig. 1) shows a slightly distorted octahedral fram of donor atoms centered by an iron atom that bears three meridional P donor groups. The angle P3-Fe1-P1 = 152.878 Å shows a significant deviation from 180°. The Fe-P distances are in the typical range of 2.18-2.22 Å.[131] Fe1-P2 distance is lengthened due to trans influence of the carbon donor.N1-Fe1 and C1-Fe1 distance s are also in typical ranges [27]. The Fe1-H1 1.480(19) Å lies in the expected range of a coordinated H to an Fe atom [28,29].

Figure 1: Molecular structure of 1. Selected bond lengths (Å) and angles(°): Fe1-H1 1.480(19), Fe1-N1 1.9434(13), Fe1-C1 1.9860(15), Fe1-P3 2.1803(5),Fe1-P1 2.1874(5),Fe1-P2 2.2146(5), N1-C7 1.314(2); N1-Fe1-C1 79.0(6); N1-Fe1-H1 172.8(7), P3-Fe-P1 152.878(18), P3-Fe1-P2 95.320(19), P1-Fe1-P2 95.66(2), N1-Fe1-P3 103.01(4), C1-Fe1-P2 167.36(5).
The X-ray diffraction study on a single crystal of 2 (Fig. 2) confirms the structure proposed in eq. 1. The iron atom is coordinated in a distorted octahedral fashion with two trimethylphosphines in trans positions P1-Fe1-P1= 153.16(3)º, An ideal equatorial plane is formed by the atoms C1 and N1 of the ortho-metalated imine coordinating with the iron atom to form a five-membered ring with a bite angle N1-Fe1-C1 = 78.93(9)°.and with a sum of internal angles (539.9º) close to the ideal value (540°) is planar. The hydrido-iron (II) complex 1 is good candidates for oxidative addition reactions. Potential ligands that do not have a lone pair of filled π type orbitals are still able to interact with transition metal complexes by breaking a σ bond. Using iodomethane as a substrate, the interesting question arises to whether attack by the metal would proceed as a regioselective addition or promote ring opening through a subsequent reductive C, C- coupling reaction. 95.705(18). Reaction of 1 in pentane proceeds as described in [Eq.2].

Figure 2: Molecular structure of 2 ( ORTEP plot with hydrogen atoms omitted). Selected bond lengths (Å) and angles(°): Fe1-N1 1.9316(19), Fe1-C1 1.966(2), Fe1-P1 2.1739(5), Fe1-P1A 2.1739(5), Fe1-P2 2.2064(8), N1-C7 1.297(3) ; N1-Fe1-C1 78.93(9), C1-Fe1-P2 168.98(7), N1-Fe1-P1 102.105(17), C1-Fe1-P1 68.71(2), P1-Fe1-P2.

Figure 3: Molecular structure of 3. Selected bond lengths (Å) and angles(°): Fe1-C1 1.957(3), Fe1-N1 1.940(2), Fe1-I1 2.7616(5), N1-C8 1.294(4), Fe1-P1 2.2587(9), Fe1-P2 2.2350(9), Fe1-P3 2.2568(9); N1-Fe1-C1 80.05(12), C1-Fe1-I1 166.44(9), N1-Fe1-P1 88.68(8); N1-Fe1-P2 175.24(9); N1-Fe1-P3 85.93(8); N1-Fe1-I 86.64(8); C1-Fe1-P2 95.51(9).

Figure 4: Molecular structure of 4. Selected bond lengths (Å) and angles(°): Fe1-C1 2.085(4), Fe1- C2 1.987(4), Fe1- N1 2.014(3), N1-C8 1.290(5), Fe1-P1 2.2241(11), Fe1-P2 2.2568(11), Fe1-P3 2.2037(11); N1-Fe1-C1 169.91(16), N1-Fe1-C2 80.34(13), C2-Fe1-P2 176.97(11), P1-Fe1-P3 168.04(4), N1-Fe1-P1 93.76(9), C1- Fe1P3 85.08(15).

Table 1: Crystal data for compound 1.

Table 2: Crystal data for compound 2.

Table 3: Crystal data for compound 3.

Table 4: Crystal data for compound 4.
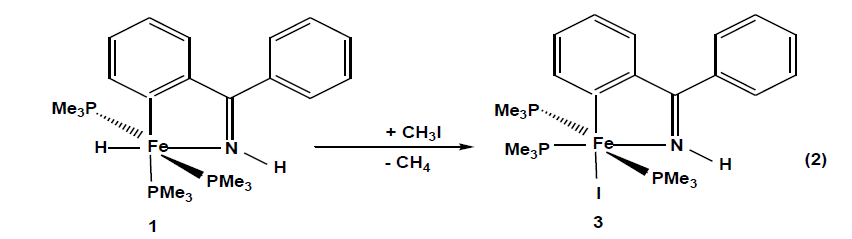
When iodomethane is added to hydrido-iron (II) complexes at-70°C. Compound 3 crystallize from pentane as dark pink cubes yield (77%) and decompose at < 143 ºC. The complete transformation of 1 into the iodo complex 3 can be monitored in the IR spectrum by the disappearance of the Fe-H band at 1730 cm-1 and there is a hypsochromic shift in all characteristic bands. In the 1H NMR spectra, there is no Fe-H resonance at high field and trimethylphosphine 1H and 13C resonances are shifted to low field. Phosphorus nuclei trans to each other resonance around 1.1 ppm and the third around 1.6 ppm. The observed patterns in the 31P NMR spectra are consistent with a meridional arrangement of the three trimethylphosphines. On the basis of chemical shifts, the electron density at the iron nucleus is subject to the σ-electron withdrawing power of the halides [30].
A molecule of the compound 3 is an octahedral geometry, a meridional arrangement of the three trimethylphosphines is recognized. The remaining three coordination sites are occupied by an iodide and a chelating imine ligand. The iodide and one trimethylphosphine P-donor are coplanar with the metallacycles forming an angle P2-Fe1-I1 97.87(3)°. Because of its weak trans influence the iodide is coordinated trans to the metalated carbon C1-Fe-I1 166.44(9)°. The five-membered metallacycle has a bite angle of 80.05(12)° and the sum of internal angles(539.85°) approach the ideal value of a planar five- membered ring. Complex 3 is distorted than 1 because iodine ligand restricts the flexibility of the trimethylphosphine groups. N1-Fe1 and C1-Fe1 distances are 1.940(2) Å and 1.957(3)Å, respectively and fall in the expected range.
The Fe1-I1 distance 2.7616(5) Å agrees with the Fe-I 2.6646 Å distance in an ortho-metalated octahedral Fe(II) compound Fe(CO)2{P(OPh)3}{(PhO)2POC6H4}(I) [31]. The two mutually trans trimethylphosphine groups have long Fe-P distances compared with the one that is trans influence of trialkylphosphines. All bond lengths are wellmatched with the other structural features [32,33].
Benzylic imines(N-methyl) react with Fe(CH3)2(PMe3)4 in THF at -70°C through the elimination of methane to afford the hexacoordinate iron(II) complex 4 [Eq. 3].
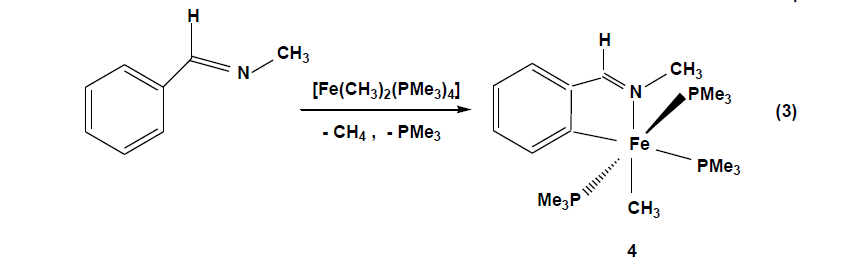
The violet solution of compound 4 in pentane deposit crystals at -27°C. The solids have moderate airsensitivity and under argon decompose at 115°C, elemental composition shows that the loss of methane and one of the trimethylphosphine groups during the reaction. The infrared spectrum of compound 4 shows the absorption band ν (C=N) of coordinated imine ligand at 1572 cm-1which occur at significantly lower energies than the free imine C=N stretching absorption, the (ν C=N,1651 cm-1). The 1H NMR spectrum of 4 in D8THF exhibits a broad singlet resonance at 1.38 ppm due to the metal-coordinated methyl group along with the expected resonances for the protons of the metalated phenyl ring from 6.74 ppm to 7.87 ppm. The 31P NMR spectrum shows a triplet at 7.6 ppm (2Jp, p = 35.6 Hz) and a doublet at 16.7ppm (2Jp, p = 35.6 Hz) with P-P coupling as expected for a mer configuration of 4.
The definitive characterization of 4 as an ortho-metalated imine complex is derived from an X-ray diffraction experiment on a single crystal. The coordination geometry around the iron atom octahedral with two Pdonor atoms of the occupying trans positions: P1-Fe-P3 = 168.04(4) °.The perpendicular plane is formed by the atoms C2 and N1of the ortho metalated N-methyl benzylidene imine (N1-Fe1-C2 = 80.34(13) °), defining a fivemembered ring with the iron atom, the P2 atom of the trimethylphosphine group disposed trans to C2 (C2-Fe1-P2 = 176.97(11) °) and the CH3 ligand located in opposite position to the nitrogen atom (N1-Fe1-C1 = 169.91(16) °). The Fe1-N1 bond length of 2.01(3) Å and the Fe1-C2 bond distance of 1.987(4) Å are typical for Fe-N and Fe-Caryl single bonds. The Fe-P distances range between 2.20 - 2.26 Å and are in agreement with the values previously found in complexes 1 – 3. Similar to the hydrido-iron(II) complexes, the reaction is believed to begin with coordination of the iron adduct to the nitrogen by substitution of one trimethylphosphine, bringing the metal closer to the ortho C-H bond. This chelation assistance facilitates highly selective C-H bond cleavage to give a cyclometalated iron (IV) intermediate. Reductive elimination of CH4 results in an ortho-metalated methyl-iron (II) complex with three trimethylphosphine ligands. Alkyl complexes of iron (II) are usually coordinatively saturated,18 electron species supported by cyclopentadienyl and carbonyl ligands.[34]
Transition metal alkyl complexes are of practical importance due to their role in numerous catalytic transformations such as olefin hydrogenation, hydroformylation, and polymerization [35]. It is the existence of several decomposition pathways that makes many transition metal alkyls unstable. Early efforts to prepare simple alkyls or aryls of transition metals showed that such compounds were generally unstable under ambient conditions. Despite the synthesis of [Me3Pt]4 by Pope and Peachy, it was assumed until quite recently that metal-carbon bonds are weak [36]. In fact, we now known that such M-C bonds are reasonably strong ranging 165-350 kJ mol-1. When it was found that the presence of ligands such as η-C5H5, CO or PR3 allowed the synthesis of thermally stable compounds, it was considered that the presence of such ligands provided better overlap between sp3 hybridized carbon and metal orbitals implying increased bond strength. A lacking β-decomposition pathway by choice of a Fe-CH3 group, a blocking imine ligand and a PMe3 ligand are in favor of stable compound 4. The principle governing their stability make a useful starting point in order to understand some of the most important organometallic reactions.