International Journal of Innovative Research in Science, Engineering and Technology
ISSN ONLINE(2319-8753)PRINT(2347-6710)
ISSN ONLINE(2319-8753)PRINT(2347-6710)
| Gurjaspreet Singh Department of Chemistry, Panjab University, Chandigarh, India. |
| Related article at Pubmed, Scholar Google |
Visit for more related articles at International Journal of Innovative Research in Science, Engineering and Technology
Four new organopropylsilatranes derived from benzotriazole are reported. The benzotriazolyl functionalized silanes (1 and 2) undergo transesterification reaction with triethanolamine and tris(isopropanol)amine in the presence of a base, thus forming the corresponding silatranes 3, 4, 5 and 6. All the silatranes have been characterized by elemental analysis and spectroscopic techniques [IR, NMR (1H,13C] and mass.
Keywords |
| Benzotriazole, Transesterification reaction, Organopropylsilatranes, Triethoxysilane |
INTRODUCTION |
| Coordination chemistry of silicon has grown tremendously over the last two decades due to the interest in this higher congener of carbon and its practical applications [1,2]. Compounds of silicon with coordination number greater than four have been known since the beginning of the 19th century, when Gay-Lussac first observed the formation of [SiF6]2− ion and the adduct of SiF4 with ammonia [3]. Thereafter, the attention of scientists was attracted by intramolecular complexes containing higher coordinated silicon [4,5]. Five or six coordinated silicon is obtained via intramolecular complexation of oxygen, nitrogen or sulfur centres as part of five or six membered chelate ring systems [6,7]. Silatranes are cyclic organosilicon ethers of tris(2-oxyalkyl)amines and constitute a unique class of pentacoordinated silicon compounds having distorted trigonal bipyramidal geometry with the silicon atom present at the bridgehead position [8-10]. The intramolecular transannular N→Si dative bond forms the basis of higher stability of silatranes than their open analogues i.e. trialkoxysilanes [11,12]. Organosilicon derivatives of nitrogen-containing heterocyclic compounds began to attract the attention of many investigators [13]. These studies made a valuable contribution to synthetic, theoretical, medicinal, and applied chemistry [14,15]. Among the numerous heterocyclic compounds, N-H hetrocycles like benzimidazole and other substituted imidazoles derivatives apparently play an increasingly important role as scaffolds for biologically active compounds and have found practical application as synthons in organic synthesis, organocatalysis, organometallic, and materials chemistry, etc [16]. Herein, we report new γ-propylsilatranes derived from benzatriazole as starting material. |
MATERIALS AND METHODS |
| Solvents were freshly dried according to standard procedures. All the reactions were carried out in dry nitrogen atmosphere. Benzotriazole (CDH), 3-chloropropyltriethoxysilane (Aldrich), triethanolamine (Aldrich) and tris(isopropanol)amine (Aldrich) were used as supplied. Infrared spectra were obtained as neat spectra on a Thermo Scientific NICOLET IS50 spectrophotometer. 1H and 13C NMR spectra were recorded on a Jeol and Bruker FT NMR (AL 400 MHz) spectrometer. Chemical shifts in ppm were determined relative to internal standard CDCl3/CCl4 and external standard tetramethylsilane (TMS). C, H and N analysis were obtained on a FLASH-2000 organic elemental analyzer. |
SYNTHESIS |
| General procedure for the synthesis of silatranes: Benzotriazole derived triethoxysilane (1 equivalent) was taken in previously dried two necked round bottomed flask fitted with a dean stark apparatus. This was then dissolved in anhydrous toluene. To the solution, trialkanolamine (1 equivalent) was added followed by catalytic amount of KOH. The resulting mixture was refluxed for 5 h to remove azeotropically the ethanol formed during the reaction. The solvent was evaporated under vacuum and 10 mL of hexane was added after which precipitation of silatrane occurred which was isolated after filteration. Synthesis of 1-(3-(1H-benzo[d][1,2,3]triazol-1-yl)propyl)-2,8,9-trioxa-5-aza-1-sila-bicyclo[3.3.3]undecane (3): The amounts used were – 1 (1 ml, 3.09 mmol), triethanolamine (0.41 ml, 3.09 mmol). Yield: 78%. Anal. Calcd for C15H22N4O3Si: C, 53.87; H, 6.63; N, 16.75. Found: C, 53.21; H, 6.35; N, 16.98. IR (Neat, cm-1): 1088 (vas SiâÃâ¬ÃâO) and 580 (v N→Si). 1H NMR (400 MHz, CDCl3, 25 °C) δ: 7.79 (t, J = 6.2 Hz, 2H), 7.72 – 7.63 (m, 2H), 3.73 (t, J = 5.8 Hz, 6H), 3.65(m, 2H), 2.80 (t, J = 5.8 Hz, 6H), 1.74 (m, 2H), 0.42 (m, 2H). 13C NMR (100 MHz, CDCl3, 25 °C) δ: 133.57, 132.32, 122.87, 57.62, 50.99, 41.26, 24.37, 13.48. MS: m/z (relative abundance (%), assignment): 335 (56, M + H)+, 357 (100, M + Na)+. |
| Synthesis of 1-(3-(1H-benzo[d][1,2,3]triazol-1-yl)propyl)-3,7,10-trimethyl-2,8,9-trioxa-5-aza-1-silabicyclo[ 3.3.3]undecane (4): The amounts used were – 1 (1 ml, 3.09 mmol), tris(isopropanol)amine (0.59 ml, 3.09 mmol). Yield: 73%. Anal. Calcd for C18H28N4O3Si: C, 57.42; H, 7.50; N, 14.88. Found: C, 57.21; H, 7.55; N, 14.98. IR (Neat, cm-1): 1092 (vas SiâÃâ¬ÃâO) and 583 (v N→Si). 1H NMR (400 MHz, CDCl3, 25 °C) δ: 7.73 (t, J = 6.2 Hz, 2H), 7.67 – 7.47 (m, 2H), 3.74 (m, 3H), 2.38(m, 6H), 1.76 (m, 2H), 1.06 (dt, 9H), 0.32 (m, 2H). 13C NMR (100 MHz, CDCl3, 25 °C) δ: 133.53, 122.88, 114.44, 66.76, 64.21, 58.88, 41.23, 25.92, 17.69, 13.54. MS: m/z (relative abundance (%), assignment): 376 (100, M + H)+, 399 (70, M + Na)+. |
| Synthesis of 1-(3-(2H-benzo[d][1,2,3]triazol-2-yl)propyl)-2,8,9-trioxa-5-aza-1-sila-bicyclo[3.3.3]undecane (5): The amounts used were – 2 (1 ml, 3.09 mmol), triethanolamine (0.41 ml, 3.09 mmol). Yield: 68%. Anal. Calcd for C15H22N4O3Si: C, 53.87; H, 6.63; N, 16.75. Found: C, 53.55; H, 6.26; N, 16.48. IR (Neat, cm-1): 1097 (vas SiâÃâ¬ÃâO) and 595 (v N→Si). 1H NMR (400 MHz, CDCl3, 25 °C) δ: 7.66 (t, J = 6.2 Hz, 2H), 7.59 – 7.49 (m, 2H), 3.60 (t, J = 5.8 Hz, 6H), 3.51(m, 2H), 2.67 (t, J = 5.8 Hz, 6H), 1.71 (m, 2H), 0.29 (m, 2H). 13C NMR (100 MHz, CDCl3, 25 °C) δ: 132.46, 131.21, 121.76, 56.52, 49.88, 40.16, 23.27, 12.37. MS: m/z (relative abundance (%), assignment): 335 (48, M + H)+, 357 (100, M + Na)+. Synthesis of 1-(3-(2H-benzo[d][1,2,3]triazol-2-yl)propyl)-3,7,10-trimethyl-2,8,9-trioxa-5-aza-1-silabicyclo[ 3.3.3]undecane (6): The amounts used were – 2 (1 ml, 3.09 mmol), tris(isopropanol)amine (0.59 ml, 3.09 mmol). Yield: 70%. Anal. Calcd for C18H28N4O3Si: C, 57.42; H, 7.50; N, 14.88. Found: C, 57.35; H, 7.76; N, 14.99. IR (Neat, cm-1): 1086 (vas SiâÃâ¬ÃâO) and 570 (v N→Si). 1H NMR (400 MHz, CDCl3, 25 °C) δ: 7.81 (t, J = 6.2 Hz, 2H), 7.73 – 7.56 (m, 2H), 3.78 (m, 3H), 2.46 (m, 6H), 1.80 (m, 2H), 1.11 (dt, 9H), 0.36 (m, 2H). 13C NMR (100 MHz, CDCl3, 25 °C) δ: 143.50, 131.37, 120.73, 112.29, 66.14, 63.12, 59.55, 39.11, 22.29, 17.73, 11.38. MS: m/z (relative abundance (%), assignment): 376 (66, M + H)+, 399 (100, M + Na)+. |
RESULTS AND DISCUSSION |
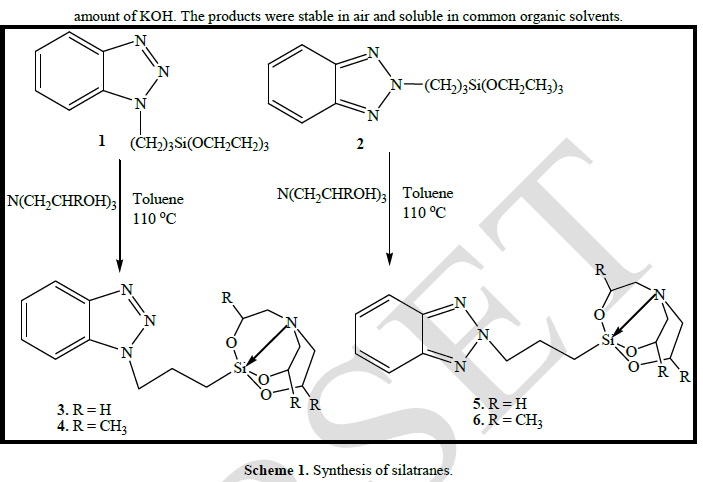
| Synthesis: As depicted in Scheme 1, the aim for the synthesis of organopropyl silatranes 3-6 was achieved by the transesterification reaction of benzotriazole derivatized triethoxysilanes with the trialkanolamine. Both triethanolamine and trisisopropanolamine were used for this purpose. The reaction proceeded smoothly in the presence of catalytic |
 |
| Characterization: |
| IR Spectroscopy: IR Spectra exhibit characteristic absorption bands of silatranyl moiety. The silatrane formation was confirmed by the observation of N→Si bond in the region 595-570 cm-1 for all compounds. The Si-O stretching vibration is assigned to the bands present in 1097-1086 cm-1. The methylene absorption bands are appeared in the range of 3010-2815 cm-1. |
| Mass spectroscopy: The mass spectra of all silatranes are consistent with the proposed structures. All the compounds have shown the common fragmentation pattern of silatranyl skeleton. |
| NMR Spectroscopy: The NMR spectroscopy (1H and 13C) data was recorded for all compounds at room temperature and results are found to be in agreement with the synthesized products. In 1H NMR specta, an upfield triplet appears for the methylene group attached to the silicon atom i.e. SiCH2 for all silatranes. Noteworthy, CCH2C proton resonances split up into a multiplet due to coupling with adjacent methylene protons and appeared in region of δ ≈ 1.45-1.60 ppm. The unsubstituted silatranes possessing Si(OCH2CH2)3N atranyl moiety consist of two intense triplet due to NCH2 (δ ≈ 2.75-2.90 ppm) and OCH2 protons (δ ≈ 3.65-3.80 ppm). For 3,7,10-trimethl substituted silatranes having N(CH2CH(O)CH3)3 moiety, multiplet and downfield shifts were appeared for CH3, CH2 and CH proton as compared to unsubstituted silatranes due to steric effects by three stereogenic C-atoms and loss of C3 symmetry. In 13C NMR spectra, the methylene carbon of propyl chain attached to silicon atom appeared as the most shielded carbon atom which is observed around δ ≈ 12.75-14.20 ppm for all silatranes. Other peak due to CCH2C is observed in region δ ≈ 24.15-25.50 ppm. The peak for CCH2N revealed large variation in region δ ≈ 42.10-50.13 ppm. All unsubstituted silatranes show two intense peaks which are assigned to silatranyl NCH2 and OCH2 respectively. Each carbon of silatranyl moiety in 3,7,10-trimethylsubstituted silatranes splits into three peaks due to steric effects. |
CONCLUSIONS |
| In this manuscript, new unsubstituted (3 and 5) and 3,7,10-trimethylsubstituted silatranes (4 and 6) possessing benzoimidazole moiety have been synthesized. All the compounds were characterized using various spectroscopic tools. The incorporation of such heterocyclic moieties in the silatranyl framework can prove to display vast advantages in the fields of material and medicinal chemistry. |
ACKNOWLEDGEMENT |
| The authors gratefully acknowledge University Grants Commission (UGC), New Delhi for providing financial support. |
References |
|