Research & Reviews in Pharmacy and Pharmaceutical Sciences
e-ISSN:2320-1215 p-ISSN: 2322-0112
e-ISSN:2320-1215 p-ISSN: 2322-0112
Meenakshi Virmani1*, and Sabir Hussain2
Department of Chemistry, Shri Jagdish Prasad Jhabarmal Tibrewala University, Jhunjhunu 333001, Churu Road, Chudela, Rajasthan, India
Department of Chemistry, Echelon Institute of Tech. (M.D University, Rohtak) Faridabad 121106, Haryana, India.
Received date: 28/02/2014 Accepted date: 18/03/2014 Revised date: 13/03/2014
Visit for more related articles at Research & Reviews in Pharmacy and Pharmaceutical Sciences
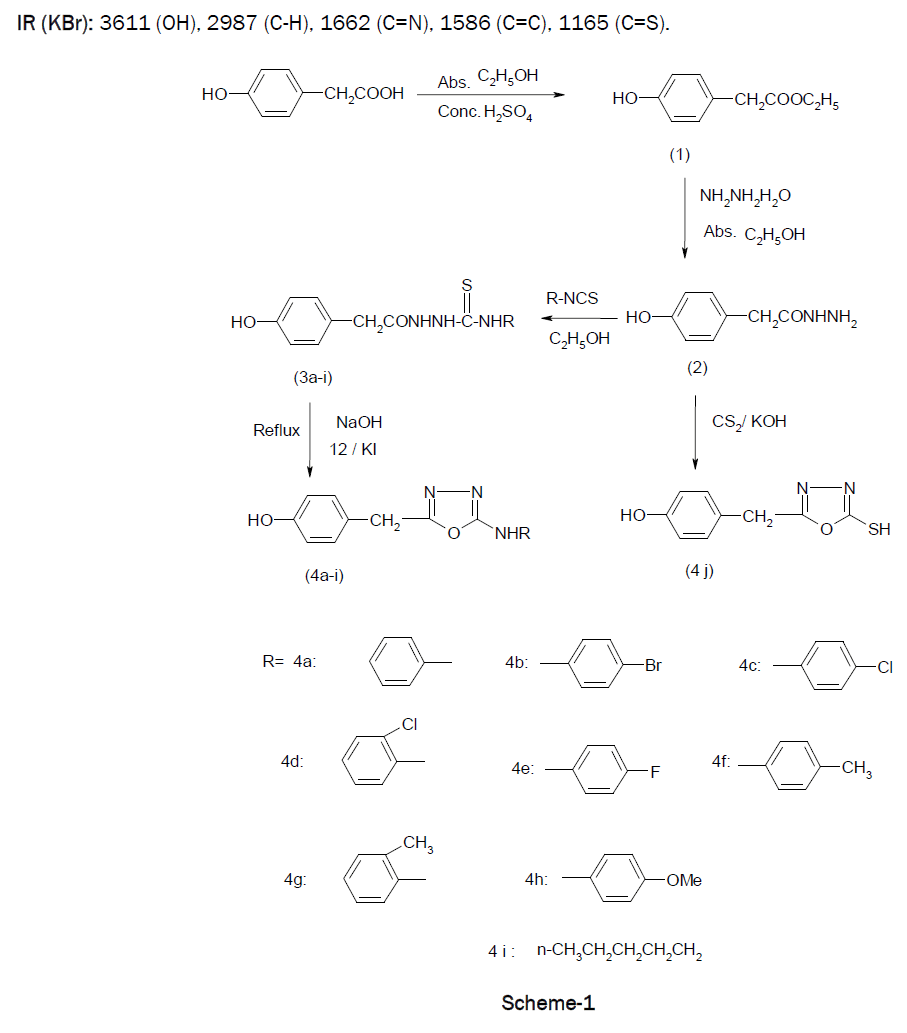
A mixture of 4-hydroxyphenylacetic acid, absolute ethanol, few drops of concentrated sulfuric acid along with a small porcelain chip on reaction formed Ethyl-(4-hydroxyphenyl) acetate (1). On condensing mixture of 1, hydrazine hydrate and absolute ethanol formed 4-Hydroxyphenyl acetic acid hydrazide (2). Further mixture of 2, aryl/alkyl isothiocyanate and ethanol was refluxed and formed N1-[2-(4-Hydroxyphenyl) acetyl] N4-alkyl/aryl-3-thiosemicarbazide (3a-i). A suspension of (3a-3i) in ethanol, aqueous sodium hydroxide, iodine in potassium iodide solution refluxed for 3-5 hours on a water bath and formed 5-(4-Hydroxyphenyl) methyl-2-phenylamino-1,3,4-oxadiazole (4a-4i). A mixture of 5, potassium hydroxide and carbon disulphide in ethanol was refluxed and formed 5-(4-Hydroxyphenyl) methyl-2-mercapto-1,3,4-oxadiazole (4j). The oxadiazole derivatives of 4-hydroxyphenyl acetic acid (4a-j) showed anti-inflammatory activity ranging from 37.37% to 66.66% at 70mg/Kg oral dose after 4 hours, whereas the standard drug Ibuprofen showed 86.35% inhibition of rat paw edema at the same oral dose.
Ibuprofen, 1,3,4-Oxadiazole Derivatives, Anti-inflammatory, Standard drug, Oxadiazole
Among a wide variety of heterocycles that have explored for developing pharmaceutically important molecules, 1,3,4 oxadiazoles constitute an important group due to their wide variety of biological activity and find wide usage as dyes, photosensitive electrical materials, polymer precursors, stabilizers, their synthesis and transformations have received great attention for a long time. Oxadiazole is a five membered heterocycle having two carbons, two nitrogen, one oxygen and two double bonds. The percentage of C, H, N present in 1, 3, 4-Oxadiazole is
Calculated % 34.29% C, 2.88% H, 40.00% N
Found % 34.56% C, 3.19% H, 39.71% N.
The IR spectra of 1, 3, 4-oxadiazole is characterized by the bonds at 1640-1560 cm-1(C=N) and 1020 cm-1(C=O). The refractive index (n25D) of 1, 3, 4-Oxadiazole is 1.43.
A number of 1,3,4 oxadiazole derivatives have been reported to show antibacterial [1], anti-inflammatory and analgesic [2], anti-proliferative [3], antimicrobial [4], antitumor [5], anti-myco-bacterial [6], anti HIV [7], Fungicidal Activities [8], Anti-Hepatitis B Virus Agents [9].
Keeping this in view, it was thought worthwhile to design the synthesis of the title compounds. The present communication reports the synthesis of unknown 5-(Hydroxyphenyl)methyl-2-alkyl/arylamino-1,3,4-oxadiazoles (4a-i) and 5-(Hydroxyphenyl) methyl-2-mercpto-1,3,4-oxadiazoles (4j) and evaluation of their anti-inflammatory activity.
To a 100 ml round bottom flask, a mixture of 4-hydroxyphenylacetic acid (0.01 mole) and absolute ethanol (50 ml) was taken. Few drops of concentrated sulphuric acid along with a small porcelain chip were added. A condenser was attached to the round bottom flask fitted with a calcium chloride guard tube to maintain anhydrous condition. The reaction mixture was refluxed for 24 hours on water bath, concentrated to give the ester. Purity of the compound was checked by TLC on silica gel G plates using toluene: ethylacetate: formic acid (5:4:1) as solvent system and the spot was located by exposure to iodine vapours. Yield: 72 %, m.p.: oily in nature, Rf : 0.76, Molecular formula: C10H12O3, Molecular weight: 180.20.
In a round bottom flask, a mixture of 1 (0.01 mole), hydrazine hydrate (0.10 mole) and absolute ethanol (50 ml) was added. A condenser with calcium chloride guard tube was attached to the flask and mixture was refluxed for 30 hours on water bath. The mixture was concentrated, cooled and poured in crushed ice. It was kept for 4-5 hours at room temperature and solid mass separated out was filtered, dried and re-crystallized from ethanol. Purity of the compound was checked by TLC on silica gel G plates using toluene: ethylacetate: formic acid (5:4:1) as solvent system and the spot was located by exposure to iodine vapours. Yield: 83 %, m.p.: 206°C, Rf : 0.15, Molecular formula: C8H10N2O2, Molecular weight: 166.18.
IR (KBr): 3596 (OH), 3406 (N-H), 2981 (C-H), 1690 (C=O), 1582 (C=C).
1HNMR (DMSO-d6): 3.20 (s, 2H, CH2), 4.19 (s, 2H, NH2), 6.65 (d, 2H, 2,6-ArH), 7.05 (d, 2H, 3,5-ArH), 9.13 (s, 1H,CONH), 9.22 (bs, 1H, OH).
A mixture of 2 (0.10 mole), aryl/alkyl isothiocyanate (0.10 mole) and ethanol (50 ml) was refluxed on water bath for 5-6 hours. It was then concentrated, cooled and kept overnight in refrigerator. White solid separated out was filtered, dried and recrystallised from ethanol acetone. Purity of the compound was checked by TLC on silica gel G plates using toluene: ethylacetate: formic acid (5:4:1) as solvent system and the spot was located by exposure to iodine vapours. The physical data of the compounds so obtained are given in Table-1.
IR (KBr): 3a-i; 3612-3571 (OH), 3392-3237 (N-H), 3028-2963 (C-H), 1696-1660 (C=O), 1589-1572 (C=C), 1183-1146 (C=S).
1HNMR (DMSO-d6): 3c; 2.50 (s, 2H, CH2), 6.67 (d, 2H, 2, 6-ArH), 7.08 (d, 2H, 3, 5-ArH), 7.37 (d, 2H, 2’, 6’-ArH-Cl), 7.46 (d, 2H, 3’, 5’-ArH-Cl), 9.27 (s, 1H, NHAr-Cl), 9.69 (bs, 2H, CONH-NH-C=S), 10.09 (bs, 1H, OH).
3h; 2.50 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 6.65 (d, 2H, 2, 6-ArH), 7.01 (d, 2H, 3, 5-ArH), 7.32 (d, 2H, 2’, 6’-ArH-OCH3), 7.45 (d, 2H, 3’, 5’-ArH-OCH3), 9.23 (s, 1H, NHAr-OCH3), 9.58 (bs, 2H, CONH-NH-C=S), 10.02 (bs, 1H, OH).

Table 1: Physical data of N1-[2-(4-Hydroxyphenyl) acetyl] N4-alkyl/aryl-3-thiosemicarbazide (3a-i)
Mass (m/z): 3c; 335 (M+), 166, 135, 107.
A suspension of 3a (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 3 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Purity of the compounds (4a-j) was checked by TLC on silica gel G plates using toluene: ethylacetate: formic acid (5:4:1) as solvent system and the spot was located by exposure to iodine vapours.
Yield: 63 %, m.p. 206°C: Rf : 0.70, Molecular formula: C15H13N3O2, Molecular weight: 267.29. %N: Found: 15.62%; Calcd: 15.72 %.
IR (KBr): 3572 (OH), 3323 (N-H), 2978 (C-H), 1664 (C=N), 1591 (C=C).
A suspension of 3b (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 4 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Yield: 69 %, m.p.: 214°C, Rf : 0.81, Molecular formula: C15H12N3O2Br, Molecular weight: 346.18. %N: Found: 11.83%; Calcd: 12.14 %.
IR (KBr): 3569 (OH), 3382 (N-H), 2988 (C-H), 1670 (C=N), 1591 (C=C), 596(C-Br).
1HNMR (DMSO-d6): 2.51 (s, 2H, CH2), 6.57 (d, 2H, 2, 6-ArH), 6.70 (d, 2H, 3, 5-ArH), 7.18 (d, 2H, 2’, 6’-ArH-Cl), 7.48 (d, 2H, 3’,5’-ArH-Cl), 9.29 (s, 1H, NH), 10.41 (s, 1H, OH).
Mass (m/z): 346 (M+), 211, 135, 107.
A suspension of 3c (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 3 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Yield: 67 %, m.p.: 222 °C, Rf : 0.70, Molecular formula: C15H12N3O2Cl, Molecular weight: 301.73. %N: Found: 13.86%; Calcd: 13.93 %.
IR (KBr): 3575 (OH), 3314 (N-H), 2960 (C-H), 1633 (C=N), 1576 (C=C), 716(C-Cl).
1HNMR (DMSO-d6): 2.50 (s, 2H, CH2), 6.56 (d, 2H, 2, 6-ArH), 6.69 (d, 2H, 3, 5-ArH), 7.23 (d, 2H, 2’, 6’-ArH-Cl), 7.53 (d, 2H, 3’, 5’-ArH-Cl), 9.33 (s, 1H, NH), 10.55 (s, 1H, OH).
Mass (m/z): 301(M+), 166, 135, 107.
A suspension of 3d (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 5 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Yield: 56 %, m.p.: 198 °C, Rf : 0.78, Molecular formula: C15H12N3O2Cl, Molecular weight: 301.73. %N: Found: 13.77%; Calcd: 13.93 %.
IR (KBr): 3577 (OH), 3359 (N-H), 2988 (C-H), 1652 (C=N), 1560 (C=C), 712 (C-Cl).
A suspension of 3e (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 4 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Yield: 62 %, m.p.242 °C, Rf: 0.70, Molecular formula: C13H12N3O2F, Molecular weight: 285.76. %N: Found: 14.58%; Calcd: 14.73 %.
IR (KBr): 3560(OH), 3300 (N-H), 2980 (C-H), 1630 (C=N), 1573 (C=C), 1090 (C-F).
A suspension of 3f (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 4 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Yield: 72 %, m.p.: 236 °C, Rf : 0.72, Molecular formula: C16H15N3O2, Molecular weight: 281.31. %N: Found: 15.03%; Calcd: 14.94 %.
IR (KBr): 3580 (OH), 3294 (N-H), 2975 (C-H), 1663 (C=N), 1575 (C=C).
1HNMR (DMSO-d6): 2.36 (s, 2H, CH3), 2.50 (s, 2H, CH2), 6.57 (d, 2H, 2, 6-ArH), 6.69 (d, 2H, 3, 5-ArH), 7.06 (d, 2H, 2’, 6’-ArH-CH3), 7.26 (d, 2H, 3’, 5’-ArH-CH3), 9.26 (s, 1H, NH), 13.25 (bs, 1H, OH).
Mass (m/z): 281 (M+), 135, 107.
A suspension of 3g (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 5 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Purity of the compound was checked by TLC on silica gel G plates using toluene: ethylacetate: formic acid (5:4:1) as solvent system and the spot was located by exposure to iodine vapours.
Yield: 64 %, m.p.: 218 °C, Rf: 0.74, Molecular formula: C16H15N3O2, Molecular weight: 281.31. %N: Found: 14.79%; Calcd: 14.94%.
IR (KBr): 3574 (OH), 3306 (N-H), 2960 (C-H), 1650 (C=N), 1571 (C=C).
A suspension of 3h (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 4 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Yield: 69 %, m.p.: 208°C, Rf: 0.68, Molecular formula: C16H15N3O3, Molecular weight: 297.31. %N: Found: 13.88%; Calcd: 14.13 %.
IR (KBr): 3569 (OH), 3336 (N-H), 2976 (C-H), 1645 (C=N), 1574 (C=C).
1HNMR (DMSO-d6): 2.50 (s, 2H, CH2), 3.80 (s, 2H, OCH3), 6.57 (d, 2H, 2, 6-ArH), 6.69 (d, 2H, 3, 5-ArH), 6.99 (d, 2H, 2’, 6’-ArH-OCH3), 7.10 (d, 2H, 3’, 5’-ArH-OCH3), 9.29 (s, 1H, NH), 13.79 (bs, 1H, OH).
A suspension of 3i (0.002 mole) in ethanol (50 ml) was dissolved in aqueous sodium hydroxide (5N, 1 ml) with cooling and stirring resulting in the formation of clear solution. To this iodine in potassium iodide solution (5%, 14 ml) was added drop wise with stirring till the colour of iodine is persisted at room temperature. The reaction mixture was refluxed for 5 hours on a water bath. It was then concentrated, kept overnight in the refrigerator and solid get separated was re-crystallized from ethanol.
Yield: 73 %, m.p. 124°C, Rf: 0.69, Molecular formula: C13H17N3O2, Molecular weight: 247.43. %N: Found: 13.26%; Calcd: 13.45 %.
IR (KBr): 3568 (OH), 3378 (N-H), 2966 (C-H), 1664 (C=N), 1573 (C=C).
1HNMR (DMSO-d6): 0.76-0.78 (t, 3H, CH3), 1.16-1.22 (merged m, 4H, CH2-CH2), 2.48 (s, 2H, CH2), 3.74-3.79 (t, 2H, NHCH2), 6.68 (d, 2H, 2, 6-ArH), 7.03 (d, 2H, 3, 5-ArH), 9.38 (s, 1H, NH), 13.53 (bs, 1H, OH).
Mass (m/z): 247 (M+), 175, 135, 107.
A mixture of hydrazide (2) (0.005 mole), potassium hydroxide (0.005 mole) and carbon disulphide (5 ml) in ethanol (50 ml) was refluxed on water bath for 10 hours. The solution was then concentrated, cooled and acidified with dilute hydrochloric acid. The solid that separated out was filtered and re-crystallized from ethanol.
Yield: 45 %, m.p. 166°C, Rf: 0.54, Molecular formula: C9H8N2O2S, Molecular weight: 208.23. %N: Found: 13.19%; Calcd: 13.45%.
IR (KBr): 3611 (OH), 2987 (C-H), 1662 (C=N), 1586 (C=C), 1165 (C=S).
The purity of the compound was checked by TLC and its characterization on the basis of IR and NMR spectral data. The IR spectrum of the compound showed peaks at 3596 cm-1, OH stretching; 3406 cm-1, NH stretching; 2981 cm-1, CH stretching; 1690 cm-1, C=O stretching and 1582 cm-1, C=C stretching vibrations of aromatic rings. The structure of the compound was further supported by its NMR spectral data which showed two singlet at δ; 3.20 and δ; 4.19 for CH2 and NH2 protons respectively. In the aromatic region two doublets centered at δ; 6.65 and δ; 7.05 were observed indicating the presence of 2,6- and 3,5- aromatic protons respectively. A singlet was observed at δ; 9.13 indicating the presence of CONH proton. Furthermore the phenolic OH proton was observed as broad singlet at δ; 9.22.
The purity of the compounds was checked by TLC and its characterization on the basis of IR, NMR and Mass spectral data. The IR spectrum of the compound (3a-i) showed peaks at 3612-3571 cm-1, OH stretching; 3392-3237 cm-1, NH stretching; 3028-2963 cm-1, CH stretching; 1695-1660 cm-1, C=O stretching and 1183-1146 cm-1, C=S stretching vibrations. The NMR spectrum of the compound 6c showed a singlet at δ 2.50 for CH2 protons. Two broad singlet were observed at δ 9.69 and δ 10.09 indicating the presence of CONH-NH-C=S and OH proton respectively. The other NH attached to p-chlorophenyl ring was also observed as a singlet at δ 9.27. In the aromatic region two doublets centered at δ 6.67 and δ 7.08 were observed indicating the presence of 2,6- and 3,5- phenolic protons respectively. The four protons of p-chlorophenyl ring were also observed as doublets centered at δ 7.37 and δ 7.46 indicating the presence of 2’,6’- and 3’,5’- aromatic protons. The structure of the compound was further supported by its mass spectral data, which showed molecular ion peak M+ at m/z 335, corresponding to the molecular formula C15H14N3O2SCl. Further peaks were observed at m/z 166, 135 and 107.
The NMR spectrum of the compound 3h showed two singlets at δ 2.50 and δ 3.81 for CH2 and OCH3 protons respectively. The signals of CONH-NH-C=S protons was observed as a broad singlet at δ 9.58, whereas the signal of OH proton was observed at δ 10.02 as a broad singlet. The NH proton attached to p-methoxyphenyl ring was also observed as a singlet at δ 9.23. In the aromatic region two doublets centered at δ 6.65 and δ 7.01 were observed indicating the presence of 2,6- and 3,5- phenolic protons respectively. The four protons of p-methoxy phenyl ring were also observed as doublets centered at δ 7.32 and δ 7.45 indicating the presence of 2’,6’- and 3’,5’- aromatic protons.
The purity of the compounds was checked by TLC and its characterization on the basis of IR, NMR and Mass spectral data. The IR spectrum of the compound (4a-i) showed peaks at 3580-3560 cm-1, OH stretching; 3382-3294 cm-1, NH stretching; 2988-2960 cm-1, CH stretching; 1670-1630 cm-1, C=N stretching and 1591-1560 cm-1, C=C stretching vibrations of aromatic rings.
The NMR spectrum of the compound 4b, 4c, 4i showed a singlet at respectively δ 2.51, δ 2.50, δ 2.48 for CH2 protons. 4f showed two singlets at δ 2.36 and δ 2.50 for CH3 and CH2 protons respectively. 4h showed two singlets at δ 2.50 and δ 3.80 for CH2 and OCH3 protons respectively.
Compounds 4b, 4c, 4f, 4h, 4i showed two doublets in the aromatic region centered respectively at δ 6.57 and δ 6.70, at δ 6.56 and δ 6.69, at δ 6.57 and δ 6.69, at δ 6.57 and δ 6.69, at δ 6.68 and δ 7.03 indicating the presence of 2, 6- and 3, 5- phenolic protons.
Compounds 4b, 4c, 4f, 4h showed four protons of p-bromophenyl ring were also observed as doublets centered respectively at δ 7.18 and δ 7.48, at δ 7.23 and δ 7.53, at δ 7.06 and δ 7.26, at δ 6.99 and δ 7.10 indicating the presence of 2’, 6’- and 3’, 5’- aromatic protons.
Compound 4b, 4c showed two singlet respectively at δ 9.29 and δ 10.41, at δ 9.33 and δ 10.55 indicating the presence of NH and OH protons. 4f, 4h, 4i showed a singlet and a broad singlet respectively at δ 9.26, δ 13.25, at δ 9.29, δ 13.79, at δ 9.38, δ 13.53 for NH and OH protons respectively.
The CH3-CH2CH2 protons of n-butyl group of 4i were merged together and obtained as a multiplet at δ 1.16-1.22. The NH-CH2 protons of n-butyl group was obtained as a triplet at δ 3.74.
The structure of the compounds 4b, 4c, 4f, 4i was further supported by its mass spectral data, which showed molecular ion peak M+ respectively at m/z 346, at m/z 301, at m/z 281, at m/z 247,corresponding to the molecular formula respectively C15H12N3O2Cl3Br 135 and 107, at m/z 135 and 107, at m/z 175, 135 and 107., C15H12N3O2Cl, C16H15N3O2, C13H17N3O2. Further peaks of 4b, 4c, 4f, 4i were observed respectively at m/z 211, 135 and 107, at m/z 166.
The purity of the compounds was checked by TLC and its characterization on the basis of IR and NMR spectral data. The IR spectrum of the compound showed peaks at 3611 cm-1, OH stretching; 2987 cm-1, CH stretching; 1662 cm-1, C=N stretching; 1586 cm-1, C=C stretching of aromatic rings and 1165 cm-1, C=S stretching vibrations.
The oxadiazole derivatives of 4-hydroxyphenyl acetic acid (4a-j) showed anti-inflammatory activity ranging from 37.37% to 66.66% at 70mg/Kg oral dose after 4 hours, whereas the standard drug Ibuprofen showed 86.35% inhibition of rat paw edema at the same oral dose (Table-2). The minimum anti-inflammatory activity was found in the compound 4a having phenyl amino group at 2nd position of the oxadiazole nucleus (37.37% inhibition). The substitution on the phenyl ring with small groups like chloro, fluoro, bromo, methyl etc. increases the activity. The compound 4h having methoxy group at the 4th position of the phenyl ring was found to be the most potent (66.66% inhibition) of the series. Replacement of the aryl amino group at 2nd position of the oxadiazole nucleus with n-butylamino group (4i) results in a compound possessing good anti-inflammatory activity (53.53% inhibition), whereas compound with mercapto group (4j) at the 2nd position showed moderate activity in comparison with the standard drug (Table-2).

Table 2: Anti-inflammatory Activity of 1,3,4-Oxadiazole Derivatives